{"title":"WHIM Syndrome: First Reported Case in a Patient of African Ancestry.","authors":"Jinal Gandhi, Michelle H Lee, Lynsie Adams, Tara Shrout Allen, Julie Li, Camille Vanessa Edwards","doi":"10.1155/2023/3888680","DOIUrl":null,"url":null,"abstract":"<p><strong>Background: </strong>Warts, hypogammaglobulinemia, infections, and myelokathexis (WHIM) syndrome is a rare, primary immunodeficiency syndrome characterized by warts, hypogammaglobulinemia, immunodeficiency, and characteristic bone marrow features of myelokathexis. The pathophysiology of WHIM syndrome is due to an autosomal dominant gain of function mutation in the CXCR4 chemokine receptor resulting in increased activity that impairs neutrophil migration from the bone marrow into the peripheral blood. This results in bone marrow distinctively crowded with mature neutrophils whose balance is shifted towards cellular senescence developing these characteristic, apoptotic nuclei termed myelokathexis. Despite the resultant severe neutropenia, the clinical syndrome is often mild and accompanied by a variety of associated abnormalities that we are just beginning to understand. <i>Case Report</i>. Diagnosis of WHIM syndrome is incredibly difficult due to phenotypic heterogeneity. To date, there are only about 105 documented cases in the scientific literature. Here, we describe the first case of WHIM syndrome documented in a patient of African ancestry. The patient in question was diagnosed at the age of 29 after a comprehensive work-up for incidental neutropenia discovered at a primary care appointment at our center in the United States. In hindsight, the patient had a history of recurrent infections, bronchiectasis, hearing loss, and VSD repair that could not be previously explained.</p><p><strong>Conclusions: </strong>Despite the challenge of timely diagnosis and the wide spectrum of clinical features that we are still discovering, WHIM syndrome tends to be a milder immunodeficiency that is highly manageable. As presented in this case, most patients respond well to G-CSF injections and newer treatments such as small-molecule CXCR4 antagonists.</p>","PeriodicalId":46307,"journal":{"name":"Case Reports in Hematology","volume":"2023 ","pages":"3888680"},"PeriodicalIF":0.7000,"publicationDate":"2023-01-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC9925260/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Case Reports in Hematology","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1155/2023/3888680","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q4","JCRName":"HEMATOLOGY","Score":null,"Total":0}
引用次数: 0
Abstract
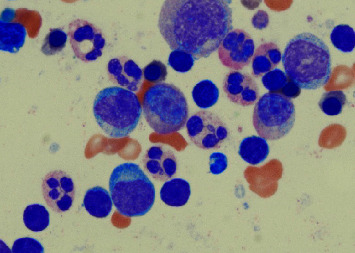
Background: Warts, hypogammaglobulinemia, infections, and myelokathexis (WHIM) syndrome is a rare, primary immunodeficiency syndrome characterized by warts, hypogammaglobulinemia, immunodeficiency, and characteristic bone marrow features of myelokathexis. The pathophysiology of WHIM syndrome is due to an autosomal dominant gain of function mutation in the CXCR4 chemokine receptor resulting in increased activity that impairs neutrophil migration from the bone marrow into the peripheral blood. This results in bone marrow distinctively crowded with mature neutrophils whose balance is shifted towards cellular senescence developing these characteristic, apoptotic nuclei termed myelokathexis. Despite the resultant severe neutropenia, the clinical syndrome is often mild and accompanied by a variety of associated abnormalities that we are just beginning to understand. Case Report. Diagnosis of WHIM syndrome is incredibly difficult due to phenotypic heterogeneity. To date, there are only about 105 documented cases in the scientific literature. Here, we describe the first case of WHIM syndrome documented in a patient of African ancestry. The patient in question was diagnosed at the age of 29 after a comprehensive work-up for incidental neutropenia discovered at a primary care appointment at our center in the United States. In hindsight, the patient had a history of recurrent infections, bronchiectasis, hearing loss, and VSD repair that could not be previously explained.
Conclusions: Despite the challenge of timely diagnosis and the wide spectrum of clinical features that we are still discovering, WHIM syndrome tends to be a milder immunodeficiency that is highly manageable. As presented in this case, most patients respond well to G-CSF injections and newer treatments such as small-molecule CXCR4 antagonists.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
