Hamid Suhail , Hongmei Peng , Jiang Xu , Hani N. Sabbah , Khalid Matrougui , Tang-Dong Liao , Pablo A. Ortiz , Kenneth E. Bernstein , Nour-Eddine Rhaleb
{"title":"Knockout of ACE-N facilitates improved cardiac function after myocardial infarction","authors":"Hamid Suhail , Hongmei Peng , Jiang Xu , Hani N. Sabbah , Khalid Matrougui , Tang-Dong Liao , Pablo A. Ortiz , Kenneth E. Bernstein , Nour-Eddine Rhaleb","doi":"10.1016/j.jmccpl.2022.100024","DOIUrl":null,"url":null,"abstract":"<div><p>Angiotensin-converting enzyme (ACE) hydrolyzes <em>N</em>-acetyl-seryl-aspartyl-lysyl-proline (Ac-SDKP) into inactive fragments through its N-terminal site (ACE<img>N). We previously showed that Ac-SDKP mediates ACE inhibitors' cardiac effects. Whether increased bioavailability of endogenous Ac-SDKP caused by knocking out ACE-N also improves cardiac function in myocardial infarction (MI)-induced heart failure (HF) is unknown. Wild-type (WT) and ACE-N knockout (ACE-NKO) mice were subjected to MI by ligating the left anterior descending artery and treated with vehicle or Ac-SDKP (1.6 mg/kg/day, s.c.) for 5 weeks, after which echocardiography was performed and left ventricles (LV) were harvested for histology and molecular biology studies. ACE-NKO mice showed increased plasma Ac-SDKP concentrations in both sham and MI group compared to WT. Exogenous Ac-SDKP further increased its circulating concentrations in WT and ACE-NKO. Shortening (SF) and ejection (EF) fractions were significantly decreased in both WT and ACE-NKO mice post-MI, but ACE-NKO mice exhibited significantly lesser decrease. Exogenous Ac-SDKP ameliorated cardiac function post-MI only in WT but failed to show any additive improvement in ACE-NKO mice. Sarcoendoplasmic reticulum calcium transport ATPase (SERCA2), a marker of cardiac function and calcium homeostasis, was significantly decreased in WT post-MI but rescued with Ac-SDKP, whereas ACE-NKO mice displayed less loss of SERCA2 expression. Our study demonstrates that gene deletion of ACE-N resulted in improved LV cardiac function in mice post-MI, which is likely mediated by increased circulating Ac-SDKP and minimally reduced expression of SERCA2. Thus, future development of specific and selective inhibitors for ACE-N could represent a novel approach to increase endogenous Ac-SDKP toward protecting the heart from post-MI remodeling.</p></div>","PeriodicalId":73835,"journal":{"name":"Journal of molecular and cellular cardiology plus","volume":"3 ","pages":"Article 100024"},"PeriodicalIF":2.2000,"publicationDate":"2023-03-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://ftp.ncbi.nlm.nih.gov/pub/pmc/oa_pdf/f0/11/nihms-1863007.PMC9910327.pdf","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of molecular and cellular cardiology plus","FirstCategoryId":"1085","ListUrlMain":"https://www.sciencedirect.com/science/article/pii/S2772976122000186","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"","JCRName":"","Score":null,"Total":0}
引用次数: 0
Abstract
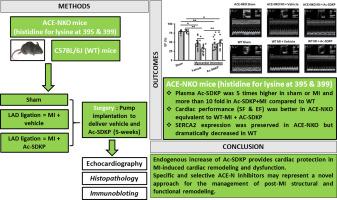
Angiotensin-converting enzyme (ACE) hydrolyzes N-acetyl-seryl-aspartyl-lysyl-proline (Ac-SDKP) into inactive fragments through its N-terminal site (ACEN). We previously showed that Ac-SDKP mediates ACE inhibitors' cardiac effects. Whether increased bioavailability of endogenous Ac-SDKP caused by knocking out ACE-N also improves cardiac function in myocardial infarction (MI)-induced heart failure (HF) is unknown. Wild-type (WT) and ACE-N knockout (ACE-NKO) mice were subjected to MI by ligating the left anterior descending artery and treated with vehicle or Ac-SDKP (1.6 mg/kg/day, s.c.) for 5 weeks, after which echocardiography was performed and left ventricles (LV) were harvested for histology and molecular biology studies. ACE-NKO mice showed increased plasma Ac-SDKP concentrations in both sham and MI group compared to WT. Exogenous Ac-SDKP further increased its circulating concentrations in WT and ACE-NKO. Shortening (SF) and ejection (EF) fractions were significantly decreased in both WT and ACE-NKO mice post-MI, but ACE-NKO mice exhibited significantly lesser decrease. Exogenous Ac-SDKP ameliorated cardiac function post-MI only in WT but failed to show any additive improvement in ACE-NKO mice. Sarcoendoplasmic reticulum calcium transport ATPase (SERCA2), a marker of cardiac function and calcium homeostasis, was significantly decreased in WT post-MI but rescued with Ac-SDKP, whereas ACE-NKO mice displayed less loss of SERCA2 expression. Our study demonstrates that gene deletion of ACE-N resulted in improved LV cardiac function in mice post-MI, which is likely mediated by increased circulating Ac-SDKP and minimally reduced expression of SERCA2. Thus, future development of specific and selective inhibitors for ACE-N could represent a novel approach to increase endogenous Ac-SDKP toward protecting the heart from post-MI remodeling.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
