Björn-Hergen Laabs, Ana Westenberger, Inke R. König
{"title":"Identification of representative trees in random forests based on a new tree-based distance measure","authors":"Björn-Hergen Laabs, Ana Westenberger, Inke R. König","doi":"10.1007/s11634-023-00537-7","DOIUrl":null,"url":null,"abstract":"<div><p>In life sciences, random forests are often used to train predictive models. However, gaining any explanatory insight into the mechanics leading to a specific outcome is rather complex, which impedes the implementation of random forests into clinical practice. By simplifying a complex ensemble of decision trees to a single most representative tree, it is assumed to be possible to observe common tree structures, the importance of specific features and variable interactions. Thus, representative trees could also help to understand interactions between genetic variants. Intuitively, representative trees are those with the minimal distance to all other trees, which requires a proper definition of the distance between two trees. Thus, we developed a new tree-based distance measure, which incorporates more of the underlying tree structure than other metrics. We compared our new method with the existing metrics in an extensive simulation study and applied it to predict the age at onset based on a set of genetic risk factors in a clinical data set. In our simulation study we were able to show the advantages of our weighted splitting variable approach. Our real data application revealed that representative trees are not only able to replicate the results from a recent genome-wide association study, but also can give additional explanations of the genetic mechanisms. Finally, we implemented all compared distance measures in R and made them publicly available in the R package timbR (https://github.com/imbs-hl/timbR).</p></div>","PeriodicalId":49270,"journal":{"name":"Advances in Data Analysis and Classification","volume":"18 2","pages":"363 - 380"},"PeriodicalIF":1.3000,"publicationDate":"2023-03-16","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://link.springer.com/content/pdf/10.1007/s11634-023-00537-7.pdf","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Advances in Data Analysis and Classification","FirstCategoryId":"94","ListUrlMain":"https://link.springer.com/article/10.1007/s11634-023-00537-7","RegionNum":4,"RegionCategory":"计算机科学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"STATISTICS & PROBABILITY","Score":null,"Total":0}
引用次数: 0
Abstract
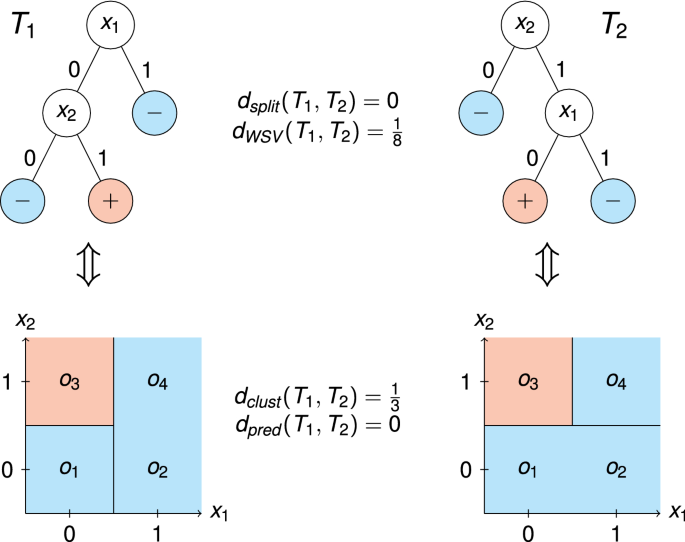
In life sciences, random forests are often used to train predictive models. However, gaining any explanatory insight into the mechanics leading to a specific outcome is rather complex, which impedes the implementation of random forests into clinical practice. By simplifying a complex ensemble of decision trees to a single most representative tree, it is assumed to be possible to observe common tree structures, the importance of specific features and variable interactions. Thus, representative trees could also help to understand interactions between genetic variants. Intuitively, representative trees are those with the minimal distance to all other trees, which requires a proper definition of the distance between two trees. Thus, we developed a new tree-based distance measure, which incorporates more of the underlying tree structure than other metrics. We compared our new method with the existing metrics in an extensive simulation study and applied it to predict the age at onset based on a set of genetic risk factors in a clinical data set. In our simulation study we were able to show the advantages of our weighted splitting variable approach. Our real data application revealed that representative trees are not only able to replicate the results from a recent genome-wide association study, but also can give additional explanations of the genetic mechanisms. Finally, we implemented all compared distance measures in R and made them publicly available in the R package timbR (https://github.com/imbs-hl/timbR).
在生命科学领域,随机森林常用于训练预测模型。然而,对导致特定结果的机理进行任何解释性洞察都相当复杂,这阻碍了随机森林在临床实践中的应用。通过将复杂的决策树组合简化为一棵最具代表性的树,我们假定可以观察到常见的树结构、特定特征的重要性以及变量之间的相互作用。因此,代表性树也有助于了解遗传变异之间的相互作用。直观地说,具有代表性的树是那些与所有其他树的距离最小的树,这就需要对两棵树之间的距离进行适当的定义。因此,我们开发了一种新的基于树的距离度量方法,与其他度量方法相比,它包含了更多的底层树结构。我们在广泛的模拟研究中将新方法与现有指标进行了比较,并将其应用于根据临床数据集中的一组遗传风险因素预测发病年龄。在模拟研究中,我们展示了加权分割变量方法的优势。我们的实际数据应用表明,代表性树不仅能够复制最近一项全基因组关联研究的结果,还能对遗传机制做出额外的解释。最后,我们用 R 语言实现了所有比较过的距离测量方法,并在 R 软件包 timbR 中公开发布(https://github.com/imbs-hl/timbR)。
期刊介绍:
The international journal Advances in Data Analysis and Classification (ADAC) is designed as a forum for high standard publications on research and applications concerning the extraction of knowable aspects from many types of data. It publishes articles on such topics as structural, quantitative, or statistical approaches for the analysis of data; advances in classification, clustering, and pattern recognition methods; strategies for modeling complex data and mining large data sets; methods for the extraction of knowledge from data, and applications of advanced methods in specific domains of practice. Articles illustrate how new domain-specific knowledge can be made available from data by skillful use of data analysis methods. The journal also publishes survey papers that outline, and illuminate the basic ideas and techniques of special approaches.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
