Intervertebral disc degeneration (IDD) is a leading contributor to low back pain (LBP). Autophagy, strongly activated by hypoxia and nutrient starvation, is a vital intracellular quality control process that removes damaged proteins and organelles to recycle them for cellular biosynthesis and energy production. While well-established as a major driver of many age-related diseases, autophagy dysregulation or deficiency has yet been confirmed to cause IDD.
In vitro, rat nucleus pulposus (NP) cells treated with bafilomycin A1 to inhibit autophagy were assessed for glycosaminoglycan (GAG) content, proteoglycan synthesis, and cell viability. In vivo, a transgenic strain (Col2a1-Cre; Atg7fl/fl) mice were successfully generated to inhibit autophagy primarily in NP tissues. Col2a1-Cre; Atg7fl/fl mouse intervertebral discs (IVDs) were evaluated for biomarkers for apoptosis and cellular senescence, aggrecan content, and histological changes up to 12 months of age.
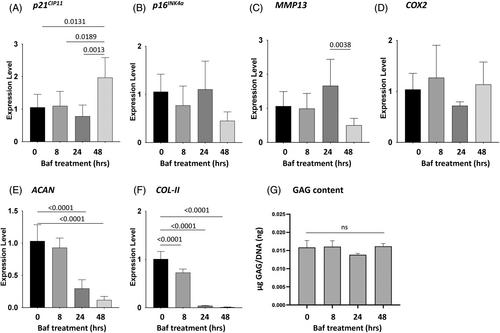
Here, we demonstrated inhibition of autophagy by bafilomycin produced IDD features in the rat NP cells, including increased apoptosis and cellular senescence (p21CIP1) and decreased expression of disc matrix genes Col2a1 and Acan. H&E histologic staining showed significant but modest degenerative changes in NP tissue of Col2a1-Cre; Atg7fl/fl mice compared to controls at 6 and 12 months of age. Intriguingly, 12-month-old Col2a1-Cre; Atg7fl/fl mice did not display increased loss of NP proteoglycan. Moreover, markers of apoptosis (cleaved caspase-3, TUNEL), and cellular senescence (p53, p16INK4a, IL-1β, TNF-α) were not affected in 12-month-old Col2a1-Cre; Atg7fl/fl mice compared to controls. However, p21CIP1and Mmp13 gene expression were upregulated in NP tissue of 12-month-old Col2a1-Cre; Atg7fl/fl mice compared to controls, suggesting p21CIP1-mediated cellular senescence resulted from NP-targeted Atg7 knockout might contribute to the observed histological changes.
The absence of overt IDD features from disrupting Atg7-mediated macroautophagy in NP tissue implicates other compensatory mechanisms, highlighting additional research needed to elucidate the complex biology of autophagy in regulating age-dependent IDD.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
