Hilal Ünsal, Canan Caka, Hacer Neslihan Bildik, Saliha Esenboğa, Alphan Kupesiz, Barış Kuşkonmaz, Duygu Uçkan Cetinkaya, Mirjam van der Burg, İlhan Tezcan, Deniz Çağdaş
{"title":"A large single‐center cohort of bare lymphocyte syndrome: Immunological and genetic features in Turkey","authors":"Hilal Ünsal, Canan Caka, Hacer Neslihan Bildik, Saliha Esenboğa, Alphan Kupesiz, Barış Kuşkonmaz, Duygu Uçkan Cetinkaya, Mirjam van der Burg, İlhan Tezcan, Deniz Çağdaş","doi":"10.1111/sji.13335","DOIUrl":null,"url":null,"abstract":"Abstract Major histocompatibility complex class II (MHC‐II) deficiency or bare lymphocyte syndrome (BLS) is a rare, early‐onset, autosomal recessive, and life‐threatening inborn error of immunity. We aimed to assess the demographic, clinical, laboratory, follow‐up, and treatment characteristics of patients with MHC‐II deficiency, together with their survival. We retrospectively investigated 21 patients with MHC‐II deficiency. Female/male ratio was 1.63. The median age at diagnosis was 16.3 months (5 months–9.7 years). Nineteen patients (90.5%) had parental consanguinity. Pulmonary diseases (pneumonia, chronic lung disease) (81%), diarrhoea (47.6%), and candidiasis (28.6%) were common. Four (19%) had autoimmunity, two developed septic arthritis, and three (14%) developed bronchiectasis in the follow‐up. Three patients (14%) had CMV viraemia, one with bilateral CMV retinitis. Eight (38.1%) had lymphocytopenia, and four (19%) had neutropenia. Serum IgM, IgA, and IgG levels were low in 18 (85.7%), 15 (71.4%), and 11 (52.4%) patients, respectively. CD4+ lymphocytopenia, a reversed CD4+/CD8+ ratio, and absent/low HLA‐DR expressions were detected in 93.3%, 86.7%, and 100% of the patients, respectively. Haematopoietic stem cell transplantation (HSCT) was performed on nine patients, and four died of septicaemia and ARDS after HSCT. The present median age of patients survived is 14 years (1–31 years). Genetic analysis was performed in 10 patients. RFX5 homozygous gene defect was found in three patients (P1, P4 and P8), and RFXANK (P2 and P14) and RFXAP (P18 and P19) heterozygous gene defects were found in each two patients, respectively. This large cohort showed that BLS patients have severe combined immunodeficiency (SCID)‐like clinical findings. Flow cytometric MHC‐II expression study is crucial for the diagnosis, differential diagnosis with SCID, early haematopoietic stem cell transplantation (HSCT), and post‐HSCT follow‐up. Genetic studies are required first for matched family donor evaluation before HSCT and then for genetic counselling.","PeriodicalId":21493,"journal":{"name":"Scandinavian Journal of Immunology","volume":"143 1","pages":"0"},"PeriodicalIF":1.6000,"publicationDate":"2023-10-15","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Scandinavian Journal of Immunology","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1111/sji.13335","RegionNum":4,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"IMMUNOLOGY","Score":null,"Total":0}
引用次数: 0
Abstract
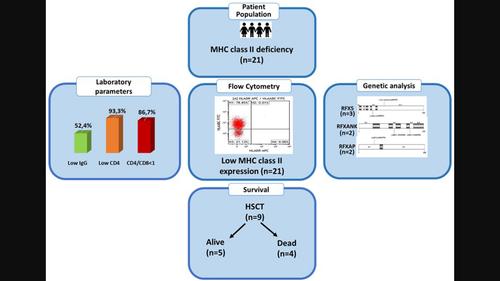
Abstract Major histocompatibility complex class II (MHC‐II) deficiency or bare lymphocyte syndrome (BLS) is a rare, early‐onset, autosomal recessive, and life‐threatening inborn error of immunity. We aimed to assess the demographic, clinical, laboratory, follow‐up, and treatment characteristics of patients with MHC‐II deficiency, together with their survival. We retrospectively investigated 21 patients with MHC‐II deficiency. Female/male ratio was 1.63. The median age at diagnosis was 16.3 months (5 months–9.7 years). Nineteen patients (90.5%) had parental consanguinity. Pulmonary diseases (pneumonia, chronic lung disease) (81%), diarrhoea (47.6%), and candidiasis (28.6%) were common. Four (19%) had autoimmunity, two developed septic arthritis, and three (14%) developed bronchiectasis in the follow‐up. Three patients (14%) had CMV viraemia, one with bilateral CMV retinitis. Eight (38.1%) had lymphocytopenia, and four (19%) had neutropenia. Serum IgM, IgA, and IgG levels were low in 18 (85.7%), 15 (71.4%), and 11 (52.4%) patients, respectively. CD4+ lymphocytopenia, a reversed CD4+/CD8+ ratio, and absent/low HLA‐DR expressions were detected in 93.3%, 86.7%, and 100% of the patients, respectively. Haematopoietic stem cell transplantation (HSCT) was performed on nine patients, and four died of septicaemia and ARDS after HSCT. The present median age of patients survived is 14 years (1–31 years). Genetic analysis was performed in 10 patients. RFX5 homozygous gene defect was found in three patients (P1, P4 and P8), and RFXANK (P2 and P14) and RFXAP (P18 and P19) heterozygous gene defects were found in each two patients, respectively. This large cohort showed that BLS patients have severe combined immunodeficiency (SCID)‐like clinical findings. Flow cytometric MHC‐II expression study is crucial for the diagnosis, differential diagnosis with SCID, early haematopoietic stem cell transplantation (HSCT), and post‐HSCT follow‐up. Genetic studies are required first for matched family donor evaluation before HSCT and then for genetic counselling.
期刊介绍:
This peer-reviewed international journal publishes original articles and reviews on all aspects of basic, translational and clinical immunology. The journal aims to provide high quality service to authors, and high quality articles for readers.
The journal accepts for publication material from investigators all over the world, which makes a significant contribution to basic, translational and clinical immunology.


 分享
分享

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
