Mauricio G. S. Costa, Mert Gur, James M. Krieger, Ivet Bahar
{"title":"Computational biophysics meets cryo-EM revolution in the search for the functional dynamics of biomolecular systems","authors":"Mauricio G. S. Costa, Mert Gur, James M. Krieger, Ivet Bahar","doi":"10.1002/wcms.1689","DOIUrl":null,"url":null,"abstract":"<p>There is a variety of experimental and computational techniques available to explore protein dynamics, each presenting advantages and limitations. One promising experimental technique that is driving the development of computational methods is cryo-electron microscopy (cryo-EM). Cryo-EM provides molecular-level structural data and first estimates of conformational landscape from single particle analysis but cannot track real-time protein dynamics and may contain uncertainties in atomic positions especially at highly dynamic regions. Molecular simulations offer atomic-level insights into protein dynamics; however, their computing time requirements limit the conformational sampling accuracy, and it is often hard, to assess by full-atomic simulations the cooperative movements of biological interest for large assemblies such as those resolved by cryo-EM. Coarse-grained (CG) simulations permit us to explore such systems, but at the costs of lower resolution and potentially incomplete sampling of conformational space. On the other hand, analytical methods may circumvent sampling limitations. In particular, elastic network models-based normal mode analyses (ENM-NMA) provide unique solutions for the complete mode spectra near equilibrium states, even for systems of megadaltons, and may thus deliver information on mechanisms of motions relevant to biological function. Yet, they lack atomic resolution as well as temporal information for non-equilibrium systems. Given the complementary nature of these methods, the integration of molecular simulations and ENM-NMA into hybrid methodologies has gained traction. This review presents the current state-of-the-art in structure-based computations and how they are helping us gain a deeper understanding of biological mechanisms, with emphasis on the development of hybrid methods accompanying the advances in cryo-EM.</p><p>This article is categorized under:\n </p>","PeriodicalId":236,"journal":{"name":"Wiley Interdisciplinary Reviews: Computational Molecular Science","volume":"14 1","pages":""},"PeriodicalIF":27.0000,"publicationDate":"2023-09-21","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://onlinelibrary.wiley.com/doi/epdf/10.1002/wcms.1689","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Wiley Interdisciplinary Reviews: Computational Molecular Science","FirstCategoryId":"92","ListUrlMain":"https://wires.onlinelibrary.wiley.com/doi/10.1002/wcms.1689","RegionNum":2,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"CHEMISTRY, MULTIDISCIPLINARY","Score":null,"Total":0}
引用次数: 0
Abstract
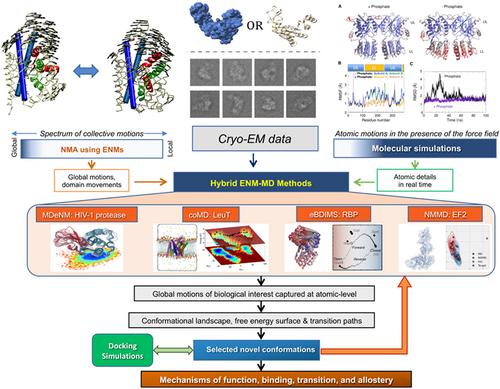
There is a variety of experimental and computational techniques available to explore protein dynamics, each presenting advantages and limitations. One promising experimental technique that is driving the development of computational methods is cryo-electron microscopy (cryo-EM). Cryo-EM provides molecular-level structural data and first estimates of conformational landscape from single particle analysis but cannot track real-time protein dynamics and may contain uncertainties in atomic positions especially at highly dynamic regions. Molecular simulations offer atomic-level insights into protein dynamics; however, their computing time requirements limit the conformational sampling accuracy, and it is often hard, to assess by full-atomic simulations the cooperative movements of biological interest for large assemblies such as those resolved by cryo-EM. Coarse-grained (CG) simulations permit us to explore such systems, but at the costs of lower resolution and potentially incomplete sampling of conformational space. On the other hand, analytical methods may circumvent sampling limitations. In particular, elastic network models-based normal mode analyses (ENM-NMA) provide unique solutions for the complete mode spectra near equilibrium states, even for systems of megadaltons, and may thus deliver information on mechanisms of motions relevant to biological function. Yet, they lack atomic resolution as well as temporal information for non-equilibrium systems. Given the complementary nature of these methods, the integration of molecular simulations and ENM-NMA into hybrid methodologies has gained traction. This review presents the current state-of-the-art in structure-based computations and how they are helping us gain a deeper understanding of biological mechanisms, with emphasis on the development of hybrid methods accompanying the advances in cryo-EM.
期刊介绍:
Computational molecular sciences harness the power of rigorous chemical and physical theories, employing computer-based modeling, specialized hardware, software development, algorithm design, and database management to explore and illuminate every facet of molecular sciences. These interdisciplinary approaches form a bridge between chemistry, biology, and materials sciences, establishing connections with adjacent application-driven fields in both chemistry and biology. WIREs Computational Molecular Science stands as a platform to comprehensively review and spotlight research from these dynamic and interconnected fields.




 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
