{"title":"Feature selection for effective prediction of SARS-COV-2 using machine learning.","authors":"Gagan Punacha, Rama Adiga","doi":"10.1007/s13258-023-01467-6","DOIUrl":null,"url":null,"abstract":"<p><strong>Background: </strong>With rise in variants of SARS-CoV-2, it is necessary to classify the emerging SARS-CoV-2 for early detection and thereby reduce human transmission. Genomic and proteomic information have less frequently been used for classifying in a machine learning (ML) approach for detection of SARS-CoV-2.</p><p><strong>Objective: </strong>With this aim we used nucleoprotein and viral proteomic evolutionary information of SARS-CoV-2 along with the charge and basicity distribution of amino acids from various strains of SARS-CoV-2 to generate a disease severity model based on ML.</p><p><strong>Methods: </strong>All sequence and clinical data were obtained from GISAID. Proteomic level calculations were added to comprise the dataset. The training set was used for feature selection. Select K- Best feature selection method was employed which was cross validated with testing set and performance evaluated. Delong's test was also done. We also employed BIRCH clustering on SARS-CoV-2 for clustering the strains.</p><p><strong>Results: </strong>Out of six ML models four were successful in training and testing. Extra Trees algorithm generated a micro-averaged F1-score of 74.2% and a weighted averaged area under the receiver operating characteristic curve (AUC-ROC) score of 73.7% with multi-class option. The feature selection set to 5, enhanced the ROC AUC from 73.7 to 76.4%. Accuracy of the selected model of 86.9% was achieved.</p><p><strong>Conclusion: </strong>The unique features identified in the ML approach was able to classify disease severity into classes and had potential for predicting risk in newer variants.</p>","PeriodicalId":12675,"journal":{"name":"Genes & genomics","volume":" ","pages":"341-354"},"PeriodicalIF":1.7000,"publicationDate":"2024-03-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Genes & genomics","FirstCategoryId":"99","ListUrlMain":"https://doi.org/10.1007/s13258-023-01467-6","RegionNum":4,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2023/11/20 0:00:00","PubModel":"Epub","JCR":"Q4","JCRName":"BIOCHEMISTRY & MOLECULAR BIOLOGY","Score":null,"Total":0}
引用次数: 0
Abstract
Background: With rise in variants of SARS-CoV-2, it is necessary to classify the emerging SARS-CoV-2 for early detection and thereby reduce human transmission. Genomic and proteomic information have less frequently been used for classifying in a machine learning (ML) approach for detection of SARS-CoV-2.
Objective: With this aim we used nucleoprotein and viral proteomic evolutionary information of SARS-CoV-2 along with the charge and basicity distribution of amino acids from various strains of SARS-CoV-2 to generate a disease severity model based on ML.
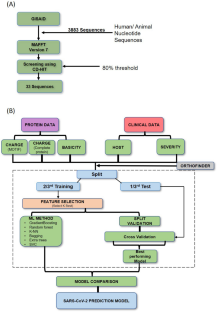
Methods: All sequence and clinical data were obtained from GISAID. Proteomic level calculations were added to comprise the dataset. The training set was used for feature selection. Select K- Best feature selection method was employed which was cross validated with testing set and performance evaluated. Delong's test was also done. We also employed BIRCH clustering on SARS-CoV-2 for clustering the strains.
Results: Out of six ML models four were successful in training and testing. Extra Trees algorithm generated a micro-averaged F1-score of 74.2% and a weighted averaged area under the receiver operating characteristic curve (AUC-ROC) score of 73.7% with multi-class option. The feature selection set to 5, enhanced the ROC AUC from 73.7 to 76.4%. Accuracy of the selected model of 86.9% was achieved.
Conclusion: The unique features identified in the ML approach was able to classify disease severity into classes and had potential for predicting risk in newer variants.
期刊介绍:
Genes & Genomics is an official journal of the Korean Genetics Society (http://kgenetics.or.kr/). Although it is an official publication of the Genetics Society of Korea, membership of the Society is not required for contributors. It is a peer-reviewed international journal publishing print (ISSN 1976-9571) and online version (E-ISSN 2092-9293). It covers all disciplines of genetics and genomics from prokaryotes to eukaryotes from fundamental heredity to molecular aspects. The articles can be reviews, research articles, and short communications.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
