Lucia Chica-Redecillas, Sergio Cuenca-Lopez, Eduardo Andres-Leon, Laura Carmen Terron-Camero, Blanca Cano-Gutierrez, Jose Manuel Cozar, Jose Antonio Lorente, Fernando Vazquez-Alonso, Luis Javier Martinez-Gonzalez, Maria Jesus Alvarez-Cubero
{"title":"Multi-omic study to unmask genes involved in prostate cancer development in a multi-case family","authors":"Lucia Chica-Redecillas, Sergio Cuenca-Lopez, Eduardo Andres-Leon, Laura Carmen Terron-Camero, Blanca Cano-Gutierrez, Jose Manuel Cozar, Jose Antonio Lorente, Fernando Vazquez-Alonso, Luis Javier Martinez-Gonzalez, Maria Jesus Alvarez-Cubero","doi":"10.1002/cac2.12501","DOIUrl":null,"url":null,"abstract":"<p>Dear Editor,</p><p>Hereditary prostate cancer (PC) comprises 5%-10% of all PC cases. The increased risk of PC in men with a family history of the disease is well known and is commonly caused by germline mutations, leading to clinical guidelines mentioning various genes for identifying high-risk individuals. However, the complex inheritance patterns involving multiple single nucleotide polymorphisms (SNPs) make it a genetically heterogeneous disease, with genetic testing still in its early stages. Current guidelines, such as those from the National Comprehensive Cancer Network (NCCN), are insufficient to identify and stratify all PC patients [<span>1</span>]. To improve testing and screening for familial PC, we report a multi-omic analysis (Supplementary Figures S1-S2) in a PC multi-case family of seven members (two healthy, four PC, and one breast cancer) (Figure 1A, Supplementary Table S1) combining exome, transcriptome and epigenomic analyses (whole-DNA methylation and small-RNA sequencing), offering a unique perspective on the understanding of hereditary PC to date. Each family is a small genetic unit that differs significantly from others with the same pathology but different genetic origins. Therefore, individualized studies may be the key to unravel the heterogeneity of this disease. However, we need to consider that conducting futuremetabolomic analysis would be next steps to reinforce present data, as well as reproducible analysis in other PC families.</p><p>We selected 34 genes based on NCCN (v1.2023) and European Association of Urology (EAU, v2.0) clinical guidelines and literature [<span>2, 3</span>] (Supplementary Table S2). We found 268 variants in 26 of these genes (<i>APC, ATM, AXIN2, BARD1, BMPR1A, BRCA1/2, CDH1, CDK4, CHEK2, DICER1, MLH1, MSH2/3/6, MUTYH, NF1, PMS2, POLD1, POLE, PTEN, RAD51C/D, SMAD4, STK11</i> and <i>TP53</i>), most of which were intronic (91.4%) and/or unreported (84.3%) (Supplementary Figure S3 and Supplementary Table S3). In addition, genome-wide analysis of high-impact variants revealed only four mutations affecting the major isoforms of the <i>ANAPC1</i>, <i>HIBCH</i>, and <i>MOK</i>, but none of these genes have been previously reported in PC (Supplementary Table S4). Interestingly, despite being high-risk cancer patients, the individuals in the present study's family did not show any pathogenic mutations in the genes specified by clinical guidelines. Furthermore, this is added to the growing evidence for the potential of non-coding mutations, both near-exonic and deep-intronic mutations, in carcinogenesis. There is already evidence of how known tumor suppressor genes are affected by intronic mutations [<span>4</span>]. Exome analysis also reported ten identical mutations in three genes, one in <i>AXIN2</i>, two in <i>DICER1</i> and seven in <i>BARD1</i>, in all PC patients (Supplementary Table S3), suggesting that these mutations may be responsible for the development of cancer in this family. Among these ten identical mutations, three in <i>BARD1</i> (c.2001+66A>C, c.1811-69T>C, and c.1811-77A>G) and one in <i>AXIN2</i> (c.-116-1330C>G) stood out with a frequency less than 0.05 in the European population. Next, we mainly focused on novel genetic markers interacting with the β-catenin pathway, <i>AXIN2</i> and <i>DICER1</i>, although other relevant data are also mentioned.</p><p><i>AXIN2</i> and <i>APC</i> have important roles in the Wnt signaling pathway as part of the β-catenin destruction complex. Partial or complete loss of these genes' activities can lead to increased β-catenin activity, resulting in aberrant activation of target genes, promoting cell proliferation and survival (Figure 1B) [<span>5</span>]. Recently, <i>DICER1</i> has also been proposed as a target gene for β-catenin. Furthermore, in liver tumors, mutations in <i>DICER1</i> have been associated with mutations in β-catenin, leading to its activation. The co-occurrence of mutations in these two genes had also been observed in endometrioid carcinoma and well-differentiated fetal lung adenocarcinoma [<span>6</span>]. Based on this scientific background, we found an identical β-catenin mutation (c.*13-8742G>A) in present cancer patients studied. This intronic mutation was found in the nonsense-mediated decay isoform (<i>CTNNB1-212</i>) of this gene and could affect both its regulation and quality control against transcriptional errors [<span>7</span>]. Activation of the Wnt signaling by mutations in <i>APC</i> or β-catenin has been observed in several cancer types and in up to 22% of castration-resistant PC [<span>8</span>].</p><p>All the aforementioned findings are supported by transcriptome and epigenetic analyses. The transcriptome study showed significant overexpression of a target gene for β-catenin, whose expression has been associated with oncogenic transformation in PC progression, <i>ALDH1A1</i> (P-value = 2.816 × 10<sup>−5</sup>, Log2-fold change (logFC) = 1.638) [<span>9</span>] (Supplementary Table S5). Overexpression of <i>KIFC1, RRM2</i> and <i>CYP1B1</i> has been observed to activate the Wnt signaling pathway. On the other hand, other cancer studies have shown that alterations in the Wnt signaling pathway co-occurred with alterations in the expression of <i>HLA-DQB2</i>, <i>CEACAM6, PTPN12, RGS18, VCAN, JMJD6</i> and <i>SOCS3</i>. These genes were differentially expressed (DE) in the sibling samples, and epigenetic analyses indicated that these changes were influenced by DNA methylation patterns and/or miRNAs. Further details about the epigenetic study can be found in Supplementary Tables S6-S7 (miRNA-mRNA integration) and Supplementary Tables S8-S9 (Methylation quantitative trait locus analysis). Additionally, the integration study revealed that <i>STK11</i>, <i>AXIN2</i> and <i>APC</i> are interconnected based on the Search Tool for the Retrieval of Interacting Genes/Proteins (STRING) database (Supplementary Figure S4 and Supplementary Table S10). <i>STK11</i> underexpression enhances WNT/β-catenin, promoting tumor progression in cholangiocarcinoma [<span>10</span>]. Enrichment analysis, predicted by the Kyoto Encyclopedia of Genes and Genomes (KEGG) and performed on the DE miRNAs according to their P-value < 0.05 and logFC > 1.3, associated eight miRNAs (hsa-miR-4443, 136-5p, 539-5p, 582-5p, 889-5p, 221-3p, 432-5p and 2115-5p) with the Wnt signaling pathway (Supplementary Figure S5 and Supplementary Table S11). Remarkably, none of the miRNAs mentioned above have been reported to date in PC. This result supports the findings from exome analysis and emphasizes the importance of intronic mutations in genes involved in the aberrant activation of this pathway. When increased cell survival is accompanied by low efficiency of cell cycle control genes in response to DNA damage, the ideal environment for cancer development is created. Thus, we also focused on highlighting the most frequently mutated gene, <i>BARD1</i> (12.31%), in the present cohort (Supplementary Figure S3) and on previously published data [<span>2</span>]. This last finding, together with the above-mentioned aspects, forms the linchpin for discerning the origin of this case of familial cancer.</p><p>Overall, the application of various omics approaches has also revealed dysregulated pathways typically observed in cancer. The DE genes identified are closely associated with immune system activation (Supplementary Figures S6-S8 and Supplementary Tables S12-S13), while the miRNAs reported are linked to PI3K-AKT-mTOR and FoxO pathways, as well as certain cancer types (Supplementary Figure S5 and S9). Furthermore, differentially methylated loci demonstrated a highly significant association with pathways related to cell membrane components, cell adhesion, metabolism and transport regulation (Supplementary Figure S10 and Supplementary Table S14). Finally, the integration of all the data showed significant correlation with immune system activation (Supplementary Figures S4 and S11 and Supplementary Tables S15-S19).</p><p>In conclusion, targeting the activation of the Wnt signaling pathway could improve the classification of inherited forms of PC. These findings suggest the importance of including <i>DICER1</i> and <i>AXIN2</i> in the gene panel of clinical guidelines for familial PC, and deeper analysis of other PC families is needed to reinforce these preliminary data. The high prevalence of previously unreported intronic mutations in these genes underscores the significance of studying non-coding regions and including them in the genetic analysis of PC. We also highlight the importance of including these above-described genes for improving the identification of individuals at risk of cancer; it will allow the development of effective prevention and treatment strategies.</p><p>Luis Javier Martinez-Gonzalez, Maria Jesus Alverez-Cubero, Jose Manuel Cozar and Jose Antonio Lorente designed the study. Blanca Cano-Gutierrez, Jose Manuel Cozar and Fernando Vazquez-Alonso treated patients. Eduardo Andres-Leon, Laura Carmen Terron-Camero, Lucia Chica-Redecillas and Sergio Cuenca analyzed and interpreted data. Eduardo Andres-Leon, Laura Carmen Terron-Camero and Lucia Chica-Redecillas performed the statistical analysis. Luis Javier Martinez-Gonzalez, Maria Jesus Alverez-Cubero and Lucia Chica-Redecillas wrote the manuscript and carried out a critical revision. Luis Javier Martinez-Gonzalez and Maria Jesus Alverez-Cubero oversaw the study. Javier Martinez-Gonzalez, Maria Jesus Alverez-Cubero and Fernando Vazquez-Alonso obtained research funding.</p><p>The study was funded by the Ministerio de Ciencia e Innovación, Spain (No. PID2019-110512RA-I00 / MCIN / AEI / 10.13039/501100011033); and Fundación para la Investigación en Urología (FIU) (No. G80445661).</p><p>The study protocol was approved by the Research Ethics Committee of the Andalusian Regional Ministry of Health (No. 0166-N-19). Written informed consent was obtained from all participants in accordance with the tenets of the Declaration of Helsinki.</p><p>The authors declare that they have no competing interests.</p><p>Not applicable.</p>","PeriodicalId":9495,"journal":{"name":"Cancer Communications","volume":"44 3","pages":"443-447"},"PeriodicalIF":24.9000,"publicationDate":"2023-11-21","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://onlinelibrary.wiley.com/doi/epdf/10.1002/cac2.12501","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Cancer Communications","FirstCategoryId":"3","ListUrlMain":"https://onlinelibrary.wiley.com/doi/10.1002/cac2.12501","RegionNum":1,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"ONCOLOGY","Score":null,"Total":0}
引用次数: 0
Abstract
Dear Editor,
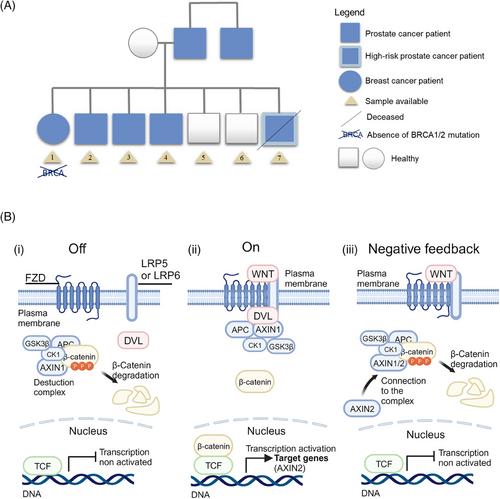
Hereditary prostate cancer (PC) comprises 5%-10% of all PC cases. The increased risk of PC in men with a family history of the disease is well known and is commonly caused by germline mutations, leading to clinical guidelines mentioning various genes for identifying high-risk individuals. However, the complex inheritance patterns involving multiple single nucleotide polymorphisms (SNPs) make it a genetically heterogeneous disease, with genetic testing still in its early stages. Current guidelines, such as those from the National Comprehensive Cancer Network (NCCN), are insufficient to identify and stratify all PC patients [1]. To improve testing and screening for familial PC, we report a multi-omic analysis (Supplementary Figures S1-S2) in a PC multi-case family of seven members (two healthy, four PC, and one breast cancer) (Figure 1A, Supplementary Table S1) combining exome, transcriptome and epigenomic analyses (whole-DNA methylation and small-RNA sequencing), offering a unique perspective on the understanding of hereditary PC to date. Each family is a small genetic unit that differs significantly from others with the same pathology but different genetic origins. Therefore, individualized studies may be the key to unravel the heterogeneity of this disease. However, we need to consider that conducting futuremetabolomic analysis would be next steps to reinforce present data, as well as reproducible analysis in other PC families.
We selected 34 genes based on NCCN (v1.2023) and European Association of Urology (EAU, v2.0) clinical guidelines and literature [2, 3] (Supplementary Table S2). We found 268 variants in 26 of these genes (APC, ATM, AXIN2, BARD1, BMPR1A, BRCA1/2, CDH1, CDK4, CHEK2, DICER1, MLH1, MSH2/3/6, MUTYH, NF1, PMS2, POLD1, POLE, PTEN, RAD51C/D, SMAD4, STK11 and TP53), most of which were intronic (91.4%) and/or unreported (84.3%) (Supplementary Figure S3 and Supplementary Table S3). In addition, genome-wide analysis of high-impact variants revealed only four mutations affecting the major isoforms of the ANAPC1, HIBCH, and MOK, but none of these genes have been previously reported in PC (Supplementary Table S4). Interestingly, despite being high-risk cancer patients, the individuals in the present study's family did not show any pathogenic mutations in the genes specified by clinical guidelines. Furthermore, this is added to the growing evidence for the potential of non-coding mutations, both near-exonic and deep-intronic mutations, in carcinogenesis. There is already evidence of how known tumor suppressor genes are affected by intronic mutations [4]. Exome analysis also reported ten identical mutations in three genes, one in AXIN2, two in DICER1 and seven in BARD1, in all PC patients (Supplementary Table S3), suggesting that these mutations may be responsible for the development of cancer in this family. Among these ten identical mutations, three in BARD1 (c.2001+66A>C, c.1811-69T>C, and c.1811-77A>G) and one in AXIN2 (c.-116-1330C>G) stood out with a frequency less than 0.05 in the European population. Next, we mainly focused on novel genetic markers interacting with the β-catenin pathway, AXIN2 and DICER1, although other relevant data are also mentioned.
AXIN2 and APC have important roles in the Wnt signaling pathway as part of the β-catenin destruction complex. Partial or complete loss of these genes' activities can lead to increased β-catenin activity, resulting in aberrant activation of target genes, promoting cell proliferation and survival (Figure 1B) [5]. Recently, DICER1 has also been proposed as a target gene for β-catenin. Furthermore, in liver tumors, mutations in DICER1 have been associated with mutations in β-catenin, leading to its activation. The co-occurrence of mutations in these two genes had also been observed in endometrioid carcinoma and well-differentiated fetal lung adenocarcinoma [6]. Based on this scientific background, we found an identical β-catenin mutation (c.*13-8742G>A) in present cancer patients studied. This intronic mutation was found in the nonsense-mediated decay isoform (CTNNB1-212) of this gene and could affect both its regulation and quality control against transcriptional errors [7]. Activation of the Wnt signaling by mutations in APC or β-catenin has been observed in several cancer types and in up to 22% of castration-resistant PC [8].
All the aforementioned findings are supported by transcriptome and epigenetic analyses. The transcriptome study showed significant overexpression of a target gene for β-catenin, whose expression has been associated with oncogenic transformation in PC progression, ALDH1A1 (P-value = 2.816 × 10−5, Log2-fold change (logFC) = 1.638) [9] (Supplementary Table S5). Overexpression of KIFC1, RRM2 and CYP1B1 has been observed to activate the Wnt signaling pathway. On the other hand, other cancer studies have shown that alterations in the Wnt signaling pathway co-occurred with alterations in the expression of HLA-DQB2, CEACAM6, PTPN12, RGS18, VCAN, JMJD6 and SOCS3. These genes were differentially expressed (DE) in the sibling samples, and epigenetic analyses indicated that these changes were influenced by DNA methylation patterns and/or miRNAs. Further details about the epigenetic study can be found in Supplementary Tables S6-S7 (miRNA-mRNA integration) and Supplementary Tables S8-S9 (Methylation quantitative trait locus analysis). Additionally, the integration study revealed that STK11, AXIN2 and APC are interconnected based on the Search Tool for the Retrieval of Interacting Genes/Proteins (STRING) database (Supplementary Figure S4 and Supplementary Table S10). STK11 underexpression enhances WNT/β-catenin, promoting tumor progression in cholangiocarcinoma [10]. Enrichment analysis, predicted by the Kyoto Encyclopedia of Genes and Genomes (KEGG) and performed on the DE miRNAs according to their P-value < 0.05 and logFC > 1.3, associated eight miRNAs (hsa-miR-4443, 136-5p, 539-5p, 582-5p, 889-5p, 221-3p, 432-5p and 2115-5p) with the Wnt signaling pathway (Supplementary Figure S5 and Supplementary Table S11). Remarkably, none of the miRNAs mentioned above have been reported to date in PC. This result supports the findings from exome analysis and emphasizes the importance of intronic mutations in genes involved in the aberrant activation of this pathway. When increased cell survival is accompanied by low efficiency of cell cycle control genes in response to DNA damage, the ideal environment for cancer development is created. Thus, we also focused on highlighting the most frequently mutated gene, BARD1 (12.31%), in the present cohort (Supplementary Figure S3) and on previously published data [2]. This last finding, together with the above-mentioned aspects, forms the linchpin for discerning the origin of this case of familial cancer.
Overall, the application of various omics approaches has also revealed dysregulated pathways typically observed in cancer. The DE genes identified are closely associated with immune system activation (Supplementary Figures S6-S8 and Supplementary Tables S12-S13), while the miRNAs reported are linked to PI3K-AKT-mTOR and FoxO pathways, as well as certain cancer types (Supplementary Figure S5 and S9). Furthermore, differentially methylated loci demonstrated a highly significant association with pathways related to cell membrane components, cell adhesion, metabolism and transport regulation (Supplementary Figure S10 and Supplementary Table S14). Finally, the integration of all the data showed significant correlation with immune system activation (Supplementary Figures S4 and S11 and Supplementary Tables S15-S19).
In conclusion, targeting the activation of the Wnt signaling pathway could improve the classification of inherited forms of PC. These findings suggest the importance of including DICER1 and AXIN2 in the gene panel of clinical guidelines for familial PC, and deeper analysis of other PC families is needed to reinforce these preliminary data. The high prevalence of previously unreported intronic mutations in these genes underscores the significance of studying non-coding regions and including them in the genetic analysis of PC. We also highlight the importance of including these above-described genes for improving the identification of individuals at risk of cancer; it will allow the development of effective prevention and treatment strategies.
Luis Javier Martinez-Gonzalez, Maria Jesus Alverez-Cubero, Jose Manuel Cozar and Jose Antonio Lorente designed the study. Blanca Cano-Gutierrez, Jose Manuel Cozar and Fernando Vazquez-Alonso treated patients. Eduardo Andres-Leon, Laura Carmen Terron-Camero, Lucia Chica-Redecillas and Sergio Cuenca analyzed and interpreted data. Eduardo Andres-Leon, Laura Carmen Terron-Camero and Lucia Chica-Redecillas performed the statistical analysis. Luis Javier Martinez-Gonzalez, Maria Jesus Alverez-Cubero and Lucia Chica-Redecillas wrote the manuscript and carried out a critical revision. Luis Javier Martinez-Gonzalez and Maria Jesus Alverez-Cubero oversaw the study. Javier Martinez-Gonzalez, Maria Jesus Alverez-Cubero and Fernando Vazquez-Alonso obtained research funding.
The study was funded by the Ministerio de Ciencia e Innovación, Spain (No. PID2019-110512RA-I00 / MCIN / AEI / 10.13039/501100011033); and Fundación para la Investigación en Urología (FIU) (No. G80445661).
The study protocol was approved by the Research Ethics Committee of the Andalusian Regional Ministry of Health (No. 0166-N-19). Written informed consent was obtained from all participants in accordance with the tenets of the Declaration of Helsinki.
The authors declare that they have no competing interests.
期刊介绍:
Cancer Communications is an open access, peer-reviewed online journal that encompasses basic, clinical, and translational cancer research. The journal welcomes submissions concerning clinical trials, epidemiology, molecular and cellular biology, and genetics.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
