{"title":"A heterozygous germline deletion within USP8 causes severe neurodevelopmental delay with multiorgan abnormalities","authors":"Masamune Sakamoto, Kenji Kurosawa, Koji Tanoue, Kazuhiro Iwama, Fumihiko Ishida, Yoshihiro Watanabe, Nobuhiko Okamoto, Naomi Tsuchida, Yuri Uchiyama, Eriko Koshimizu, Atsushi Fujita, Kazuharu Misawa, Satoko Miyatake, Takeshi Mizuguchi, Naomichi Matsumoto","doi":"10.1038/s10038-023-01209-2","DOIUrl":null,"url":null,"abstract":"Ubiquitin-specific protease 8 (USP8) is a deubiquitinating enzyme involved in deubiquitinating the enhanced epidermal growth factor receptor for escape from degradation. Somatic variants at a hotspot in USP8 are a cause of Cushing’s disease, and a de novo germline USP8 variant at this hotspot has been described only once previously, in a girl with Cushing’s disease and developmental delay. In this study, we investigated an exome-negative patient with severe developmental delay, dysmorphic features, and multiorgan dysfunction by long-read sequencing, and identified a 22-kb de novo germline deletion within USP8 (chr15:50469966-50491995 [GRCh38]). The deletion involved the variant hotspot, one rhodanese domain, and two SH3 binding motifs, and was presumed to be generated through nonallelic homologous recombination through Alu elements. Thus, the patient may have perturbation of the endosomal sorting system and mitochondrial autophagy through the USP8 defect. This is the second reported case of a germline variant in USP8.","PeriodicalId":16077,"journal":{"name":"Journal of Human Genetics","volume":"69 2","pages":"85-90"},"PeriodicalIF":2.5000,"publicationDate":"2023-11-30","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Human Genetics","FirstCategoryId":"99","ListUrlMain":"https://www.nature.com/articles/s10038-023-01209-2","RegionNum":3,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"GENETICS & HEREDITY","Score":null,"Total":0}
引用次数: 0
Abstract
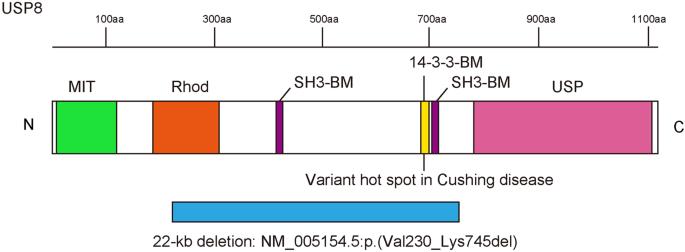
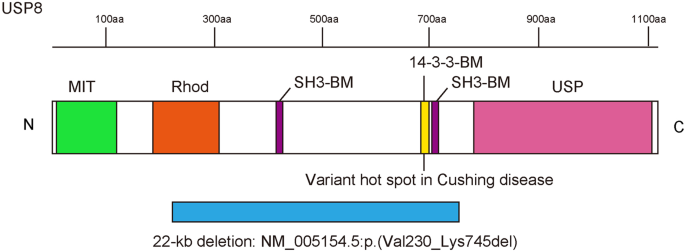
Ubiquitin-specific protease 8 (USP8) is a deubiquitinating enzyme involved in deubiquitinating the enhanced epidermal growth factor receptor for escape from degradation. Somatic variants at a hotspot in USP8 are a cause of Cushing’s disease, and a de novo germline USP8 variant at this hotspot has been described only once previously, in a girl with Cushing’s disease and developmental delay. In this study, we investigated an exome-negative patient with severe developmental delay, dysmorphic features, and multiorgan dysfunction by long-read sequencing, and identified a 22-kb de novo germline deletion within USP8 (chr15:50469966-50491995 [GRCh38]). The deletion involved the variant hotspot, one rhodanese domain, and two SH3 binding motifs, and was presumed to be generated through nonallelic homologous recombination through Alu elements. Thus, the patient may have perturbation of the endosomal sorting system and mitochondrial autophagy through the USP8 defect. This is the second reported case of a germline variant in USP8.
期刊介绍:
The Journal of Human Genetics is an international journal publishing articles on human genetics, including medical genetics and human genome analysis. It covers all aspects of human genetics, including molecular genetics, clinical genetics, behavioral genetics, immunogenetics, pharmacogenomics, population genetics, functional genomics, epigenetics, genetic counseling and gene therapy.
Articles on the following areas are especially welcome: genetic factors of monogenic and complex disorders, genome-wide association studies, genetic epidemiology, cancer genetics, personal genomics, genotype-phenotype relationships and genome diversity.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
