Programmed cell death (PCD) has been widely investigated in various human diseases. The present study aimed to identify a novel PCD-related genetic signature in cervical squamous cell carcinoma (CESC) to provide clues for survival, immunotherapy and drug sensitization prediction.
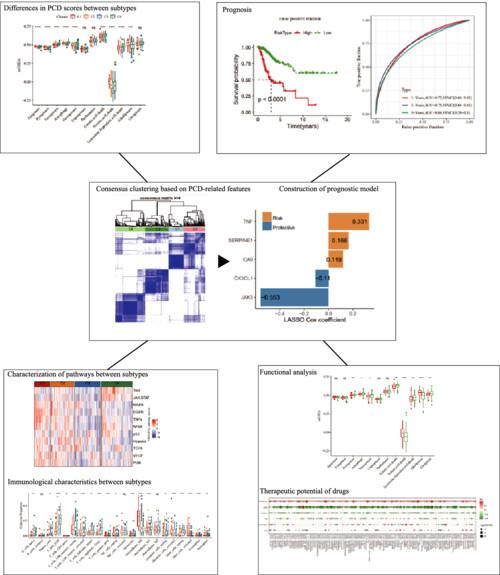
Single-sample gene set enrichment analysis (ssGSEA) was used to quantify the PCD score and assess the distribution of PCD in clinicopathological characteristics in The Cancer Genome Atlas (TCGA)-CESC samples. Then, the ConsensusClusterPlus method was used to identify molecular subtypes in the TCGA-CESC database. Genomic mutation analysis, Gene Ontology and Kyoto Encyclopedia of Genes and Genomes functional enrichment, as well as tumor microenvironment (TME) infiltration analysis, were performed for each molecular subtype group. Finally, a prognostic model by Uni-Cox and least absolute shrinkage and selection operator-Cox analysis was established based on differentially expressed genes from molecular subtypes. ESTIMATE (i.e. Estimation of STromal and Immune cells in MAlignantTumours using Expression data) and ssGSEA were performed to assess the correlation between the model and TME. Drug sensitization prediction was carried out with the oncoPredict package.
Preliminary analysis indicated that PCD had a potential association clinical characteristics of the TCGA-CESC cohort, and PCD-related genes mutated in 289 (70.59%) CESC patients. Next, four groups of CESC molecular typing were clustered based on 63 significantly prognostic PCD-related genes. Among four subtypes, C1 group displayed the worst prognosis combined with over expressed PCD genes and enriched cell cycle-related pathways. C4 group exhibited the best prognosis accompanied with high degree of immune infiltration. Finally, a five-gene (SERPINE1, TNF, CA9, CX3CL1 and JAK3) prognostic model was constructed. Patients in the high-risk group displayed unfavorable survival. Immune infiltration analysis found that the low-risk group had significantly higher levels of immune cell infiltration such as T cells, Macrophages_M1, relative to the high-risk group, and were significantly enriched in apoptosis-associated pathways, which predicted a higher level of immunity. Drug sensitivity correlation analysis revealed that the high-risk group was resistant to conventional chemotherapeutic drugs and sensitive to the Food and Drug Administration-approved drugs BI.2536_1086 and SCH772984_1564.
In the present study, we first found that PCD-related gene expression patterns were correlated with clinical features of CESC patients, which predicts the feasibility of subsequent mining of prognostic features based on these genes. The five-PCD-associated-gene prognostic model showed good assessment ability in predicting patient prognosis, immune response and drug-sensitive response, and provided guidance for the elucidation of the mechanism by which PCD affects CESC, as well as for the clinical targeting of drugs.




 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
