Severe Unilateral Microtia with Aural Atresia, Hair White Patch, Stereotypes in a Young Boy with De novo 16p13.11 Deletion: Reasons for a New Genotype-Phenotype Correlation.
Piero Pavone, Xena Giada Pappalardo, Claudia Parano, Enrico Parano, Antonio Corsello, Martino Ruggieri, Giovanni Cacciaguerra, Raffaele Falsaperla
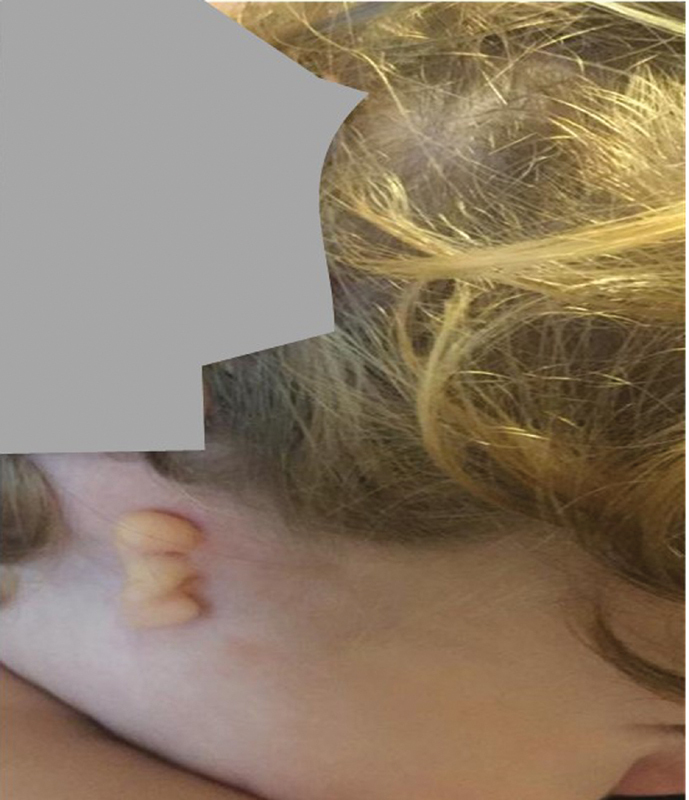
{"title":"Severe Unilateral Microtia with Aural Atresia, Hair White Patch, Stereotypes in a Young Boy with De novo 16p13.11 Deletion: Reasons for a New Genotype-Phenotype Correlation.","authors":"Piero Pavone, Xena Giada Pappalardo, Claudia Parano, Enrico Parano, Antonio Corsello, Martino Ruggieri, Giovanni Cacciaguerra, Raffaele Falsaperla","doi":"10.1055/s-0043-1777362","DOIUrl":null,"url":null,"abstract":"<p><p><b>Background</b> Microtia is an uncommon congenital malformation ranging from mild anatomic structural abnormalities to partial or complete absence of the ear leading to hearing impairment. Congenital microtia may present as a single malformation (isolated microtia) or sometimes associated with other congenital anomalies involving various organs. Microtia has been classified in three degrees according to the complexity of the auricular malformation and to anotia referred to the total absence of the ear. Genetic role in causing auricular malformation has been widely demonstrated, and genotype-phenotype correlation has been reported in cases of syndromic microtia. <b>Case Presentation</b> We report here a young patient with a third degree of scale classification and aural atresia. The patient showed unspecific facial dysmorphism, speech delay, precocious teething, hair white patch, and stereotypic anomalous movements. Genetic analysis displayed a de novo 16p13.11 deletion. <b>Conclusion</b> Microtia with aural atresia is an uncommon and severe birth defect, which affects functional and esthetic aspects, often associated with other malformations. As traumatic this disorder may be for the parents, the microtia and aural atresia are treatable, thanks to the improving and evolving surgical techniques. Based on the genetic analysis and the clinical features observed in the present case, a genotype-phenotype correlation has been proposed.</p>","PeriodicalId":40142,"journal":{"name":"Global Medical Genetics","volume":"10 4","pages":"370-375"},"PeriodicalIF":1.5000,"publicationDate":"2023-12-04","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC10695706/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Global Medical Genetics","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1055/s-0043-1777362","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2023/12/1 0:00:00","PubModel":"eCollection","JCR":"Q4","JCRName":"GENETICS & HEREDITY","Score":null,"Total":0}
引用次数: 0
Abstract
Background Microtia is an uncommon congenital malformation ranging from mild anatomic structural abnormalities to partial or complete absence of the ear leading to hearing impairment. Congenital microtia may present as a single malformation (isolated microtia) or sometimes associated with other congenital anomalies involving various organs. Microtia has been classified in three degrees according to the complexity of the auricular malformation and to anotia referred to the total absence of the ear. Genetic role in causing auricular malformation has been widely demonstrated, and genotype-phenotype correlation has been reported in cases of syndromic microtia. Case Presentation We report here a young patient with a third degree of scale classification and aural atresia. The patient showed unspecific facial dysmorphism, speech delay, precocious teething, hair white patch, and stereotypic anomalous movements. Genetic analysis displayed a de novo 16p13.11 deletion. Conclusion Microtia with aural atresia is an uncommon and severe birth defect, which affects functional and esthetic aspects, often associated with other malformations. As traumatic this disorder may be for the parents, the microtia and aural atresia are treatable, thanks to the improving and evolving surgical techniques. Based on the genetic analysis and the clinical features observed in the present case, a genotype-phenotype correlation has been proposed.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
