{"title":"Jellyfish: A modular code for wave function-based electron dynamics simulations and visualizations on traditional and quantum compute architectures","authors":"Fabian Langkabel, Pascal Krause, Annika Bande","doi":"10.1002/wcms.1696","DOIUrl":null,"url":null,"abstract":"<p>Ultrafast electron dynamics have made rapid progress in the last few years. With Jellyfish, we now introduce a program suite that enables to perform the entire workflow of an electron-dynamics simulation. The modular program architecture offers a flexible combination of different propagators, Hamiltonians, basis sets, and more. Jellyfish can be operated by a graphical user interface, which makes it easy to get started for nonspecialist users and gives experienced users a clear overview of the entire functionality. The temporal evolution of a wave function can currently be executed in the time-dependent configuration interaction method (TDCI) formalism, however, a plugin system facilitates the expansion to other methods and tools without requiring in-depth knowledge of the program. Currently developed plugins allow to include results from conventional electronic structure calculations as well as the usage and extension of quantum-compute algorithms for electron dynamics. We present the capabilities of Jellyfish on three examples to showcase the simulation and analysis of light-driven correlated electron dynamics. The implemented visualization of various densities enables an efficient and detailed analysis for the long-standing quest of the electron–hole pair formation.</p><p>This article is categorized under:\n </p>","PeriodicalId":236,"journal":{"name":"Wiley Interdisciplinary Reviews: Computational Molecular Science","volume":"14 1","pages":""},"PeriodicalIF":27.0000,"publicationDate":"2023-11-27","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://onlinelibrary.wiley.com/doi/epdf/10.1002/wcms.1696","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Wiley Interdisciplinary Reviews: Computational Molecular Science","FirstCategoryId":"92","ListUrlMain":"https://wires.onlinelibrary.wiley.com/doi/10.1002/wcms.1696","RegionNum":2,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"CHEMISTRY, MULTIDISCIPLINARY","Score":null,"Total":0}
引用次数: 0
Abstract
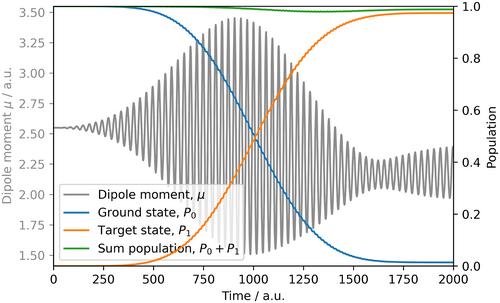
Ultrafast electron dynamics have made rapid progress in the last few years. With Jellyfish, we now introduce a program suite that enables to perform the entire workflow of an electron-dynamics simulation. The modular program architecture offers a flexible combination of different propagators, Hamiltonians, basis sets, and more. Jellyfish can be operated by a graphical user interface, which makes it easy to get started for nonspecialist users and gives experienced users a clear overview of the entire functionality. The temporal evolution of a wave function can currently be executed in the time-dependent configuration interaction method (TDCI) formalism, however, a plugin system facilitates the expansion to other methods and tools without requiring in-depth knowledge of the program. Currently developed plugins allow to include results from conventional electronic structure calculations as well as the usage and extension of quantum-compute algorithms for electron dynamics. We present the capabilities of Jellyfish on three examples to showcase the simulation and analysis of light-driven correlated electron dynamics. The implemented visualization of various densities enables an efficient and detailed analysis for the long-standing quest of the electron–hole pair formation.
期刊介绍:
Computational molecular sciences harness the power of rigorous chemical and physical theories, employing computer-based modeling, specialized hardware, software development, algorithm design, and database management to explore and illuminate every facet of molecular sciences. These interdisciplinary approaches form a bridge between chemistry, biology, and materials sciences, establishing connections with adjacent application-driven fields in both chemistry and biology. WIREs Computational Molecular Science stands as a platform to comprehensively review and spotlight research from these dynamic and interconnected fields.




 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
