The explosion of available genomic data poses significant opportunities and challenges for genome-wide association studies. Current approaches via linear mixed models (LMM) are straightforward but prevent flexible assumptions of an a priori genomic architecture, while Bayesian sparse LMMs (BSLMMs) allow this flexibility. Complex traits, such as specialized metabolites, are subject to various hierarchical effects, including gene regulation, enzyme efficiency, and the availability of reactants.
To identify alternative genetic architectures, we examined the genetic architecture underlying the carotenoid content of an association mapping panel of Helianthus annuus individuals using multiple BSLMM and LMM frameworks.
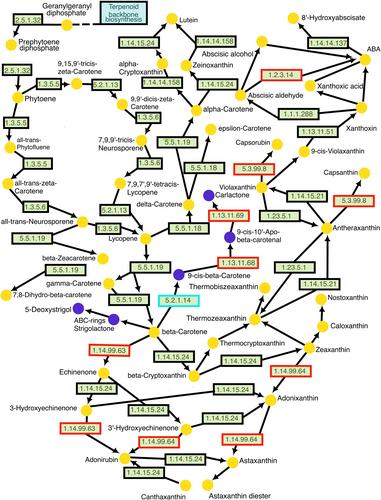
The LMMs of genome-wide single-nucleotide polymorphisms (SNPs) identified a single transcription factor responsible for the observed variations in the carotenoid content; however, a BSLMM of the SNPs with the bottom 1% of effect sizes from the results of the LMM identified multiple biologically relevant quantitative trait loci (QTLs) for carotenoid content external to the known (annotated) carotenoid pathway. A candidate pathway analysis (CPA) suggested a β-carotene isomerase to be the enzyme with the highest impact on the observed carotenoid content within the carotenoid pathway.
While traditional LMM approaches suggested a single unknown transcription factor associated with carotenoid content variation in sunflower petals, BSLMM proposed several QTLs with interpretable biological relevance to this trait. In addition, the CPA allowed for the dissection of the regulatory vs. biosynthetic genetic architectures underlying this metabolic trait.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
