Bryan L. Dinh, Echo Tang, Kekoa Taparra, Nathan Nakatsuka, Fei Chen, Charleston W. K. Chiang
{"title":"Recombination map tailored to Native Hawaiians may improve robustness of genomic scans for positive selection","authors":"Bryan L. Dinh, Echo Tang, Kekoa Taparra, Nathan Nakatsuka, Fei Chen, Charleston W. K. Chiang","doi":"10.1007/s00439-023-02625-2","DOIUrl":null,"url":null,"abstract":"<p>Recombination events establish the patterns of haplotypic structure in a population and estimates of recombination rates are used in several downstream population and statistical genetic analyses. Using suboptimal maps from distantly related populations may reduce the efficacy of genomic analyses, particularly for underrepresented populations such as the Native Hawaiians. To overcome this challenge, we constructed recombination maps using genome-wide array data from two study samples of Native Hawaiians: one reflecting the current admixed state of Native Hawaiians (NH map) and one based on individuals of enriched Polynesian ancestries (PNS map) with the potential to be used for less admixed Polynesian populations such as the Samoans. We found the recombination landscape to be less correlated with those from other continental populations (e.g. Spearman’s rho = 0.79 between PNS and CEU (Utah residents with Northern and Western European ancestry) compared to 0.92 between YRI (Yoruba in Ibadan, Nigeria) and CEU at 50 kb resolution), likely driven by the unique demographic history of the Native Hawaiians. PNS also shared the fewest recombination hotspots with other populations (e.g. 8% of hotspots shared between PNS and CEU compared to 27% of hotspots shared between YRI and CEU). We found that downstream analyses in the Native Hawaiian population, such as local ancestry inference, imputation, and IBD segment and relatedness detections, would achieve similar efficacy when using the NH map compared to an omnibus map. However, for genome scans of adaptive loci using integrated haplotype scores, we found several loci with apparent genome-wide significant signals (|<i>Z</i>-score|> 4) in Native Hawaiians that would not have been significant when analyzed using NH-specific maps. Population-specific recombination maps may therefore improve the robustness of haplotype-based statistics and help us better characterize the evolutionary history that may underlie Native Hawaiian-specific health conditions that persist today.</p>","PeriodicalId":13175,"journal":{"name":"Human Genetics","volume":"25 1","pages":""},"PeriodicalIF":3.6000,"publicationDate":"2023-12-29","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Human Genetics","FirstCategoryId":"99","ListUrlMain":"https://doi.org/10.1007/s00439-023-02625-2","RegionNum":2,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"GENETICS & HEREDITY","Score":null,"Total":0}
引用次数: 0
Abstract
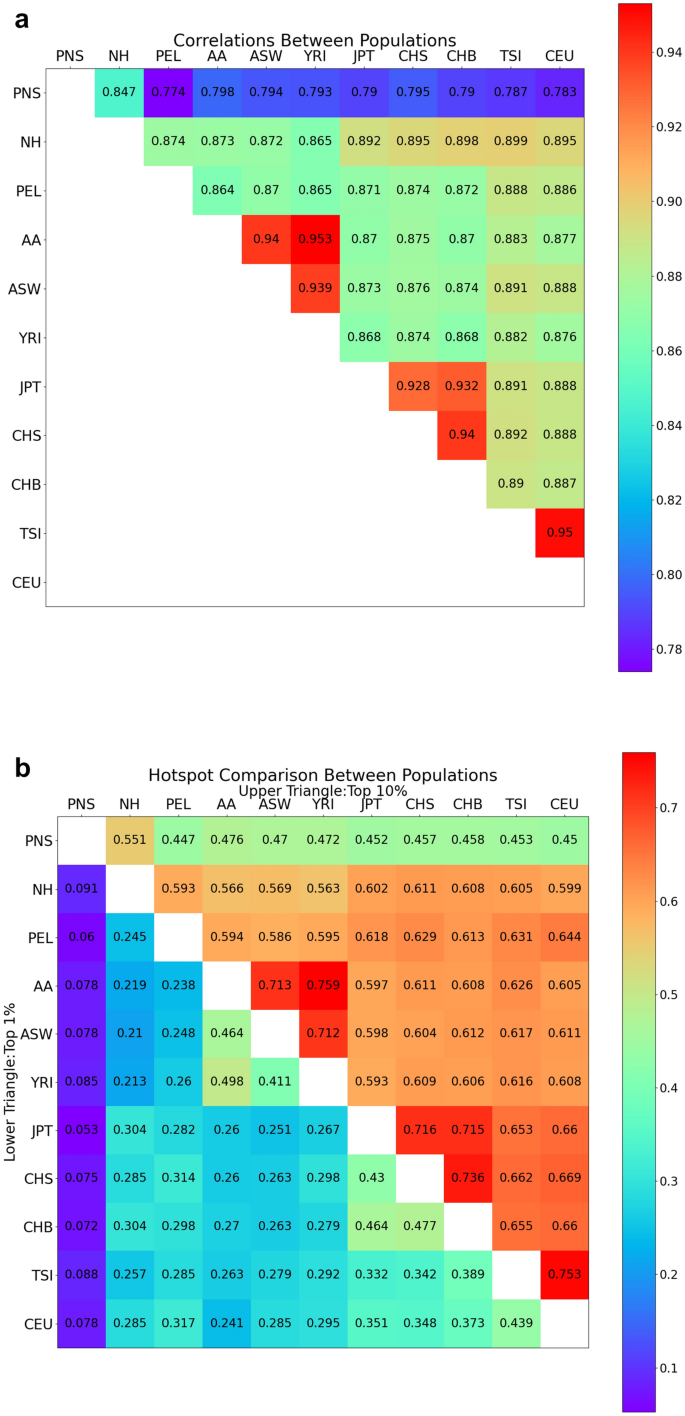
Recombination events establish the patterns of haplotypic structure in a population and estimates of recombination rates are used in several downstream population and statistical genetic analyses. Using suboptimal maps from distantly related populations may reduce the efficacy of genomic analyses, particularly for underrepresented populations such as the Native Hawaiians. To overcome this challenge, we constructed recombination maps using genome-wide array data from two study samples of Native Hawaiians: one reflecting the current admixed state of Native Hawaiians (NH map) and one based on individuals of enriched Polynesian ancestries (PNS map) with the potential to be used for less admixed Polynesian populations such as the Samoans. We found the recombination landscape to be less correlated with those from other continental populations (e.g. Spearman’s rho = 0.79 between PNS and CEU (Utah residents with Northern and Western European ancestry) compared to 0.92 between YRI (Yoruba in Ibadan, Nigeria) and CEU at 50 kb resolution), likely driven by the unique demographic history of the Native Hawaiians. PNS also shared the fewest recombination hotspots with other populations (e.g. 8% of hotspots shared between PNS and CEU compared to 27% of hotspots shared between YRI and CEU). We found that downstream analyses in the Native Hawaiian population, such as local ancestry inference, imputation, and IBD segment and relatedness detections, would achieve similar efficacy when using the NH map compared to an omnibus map. However, for genome scans of adaptive loci using integrated haplotype scores, we found several loci with apparent genome-wide significant signals (|Z-score|> 4) in Native Hawaiians that would not have been significant when analyzed using NH-specific maps. Population-specific recombination maps may therefore improve the robustness of haplotype-based statistics and help us better characterize the evolutionary history that may underlie Native Hawaiian-specific health conditions that persist today.
期刊介绍:
Human Genetics is a monthly journal publishing original and timely articles on all aspects of human genetics. The Journal particularly welcomes articles in the areas of Behavioral genetics, Bioinformatics, Cancer genetics and genomics, Cytogenetics, Developmental genetics, Disease association studies, Dysmorphology, ELSI (ethical, legal and social issues), Evolutionary genetics, Gene expression, Gene structure and organization, Genetics of complex diseases and epistatic interactions, Genetic epidemiology, Genome biology, Genome structure and organization, Genotype-phenotype relationships, Human Genomics, Immunogenetics and genomics, Linkage analysis and genetic mapping, Methods in Statistical Genetics, Molecular diagnostics, Mutation detection and analysis, Neurogenetics, Physical mapping and Population Genetics. Articles reporting animal models relevant to human biology or disease are also welcome. Preference will be given to those articles which address clinically relevant questions or which provide new insights into human biology.
Unless reporting entirely novel and unusual aspects of a topic, clinical case reports, cytogenetic case reports, papers on descriptive population genetics, articles dealing with the frequency of polymorphisms or additional mutations within genes in which numerous lesions have already been described, and papers that report meta-analyses of previously published datasets will normally not be accepted.
The Journal typically will not consider for publication manuscripts that report merely the isolation, map position, structure, and tissue expression profile of a gene of unknown function unless the gene is of particular interest or is a candidate gene involved in a human trait or disorder.




 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
