{"title":"C-terminal truncations in IQSEC2: implications for synaptic localization, guanine nucleotide exchange factor activity, and neurological manifestations","authors":"Moeko Nakashima, Tomoko Shiroshima, Masahiro Fukaya, Takeyuki Sugawara, Hiroyuki Sakagami, Kazuki Yamazawa","doi":"10.1038/s10038-023-01210-9","DOIUrl":null,"url":null,"abstract":"IQSEC2 gene on chromosome Xq11.22 encodes a member of guanine nucleotide exchange factor (GEF) protein that is implicated in the activation of ADP-ribosylation factors (Arfs) at the postsynaptic density (PSD), and plays a crucial role in synaptic transmission and dendritic spine formation. Alterations in IQSEC2 have been linked to X-linked intellectual developmental disorders including epilepsy and behavioral abnormalities. Of interest, truncating variants at the C-terminus of IQSEC2 can cause severe phenotypes, akin to truncating variants located in other regions. Here, we present a 5-year-old boy with severe intellectual disability and progressive epilepsy. The individual carried a nonsense variant p.Q1227* in the last exon of the IQSEC2 gene that was supposed to escape nonsense-mediated mRNA decay, thereby leading to a translation of C-terminus truncated IQSEC2 protein with residual activity. The functional analyses showed that the GEF activity of IQSEC2 Q1227* was compromised, and that the IQSEC2 Q1227* lacked preferential synaptic localization due to the absence of functional domains for binding to scaffolding proteins in the PSD. The impaired GEF activity and disrupted synaptic localization of the mutant IQSEC2 protein could impact dendritic and spine development in neurons, potentially explaining the patient’s severe neurological manifestations. Our findings indicate that C-terminal truncations in IQSEC2, previously not well-characterized, may have significant pathogenic implications.","PeriodicalId":16077,"journal":{"name":"Journal of Human Genetics","volume":"69 3-4","pages":"119-123"},"PeriodicalIF":2.6000,"publicationDate":"2024-01-10","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Human Genetics","FirstCategoryId":"99","ListUrlMain":"https://www.nature.com/articles/s10038-023-01210-9","RegionNum":3,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"GENETICS & HEREDITY","Score":null,"Total":0}
引用次数: 0
Abstract
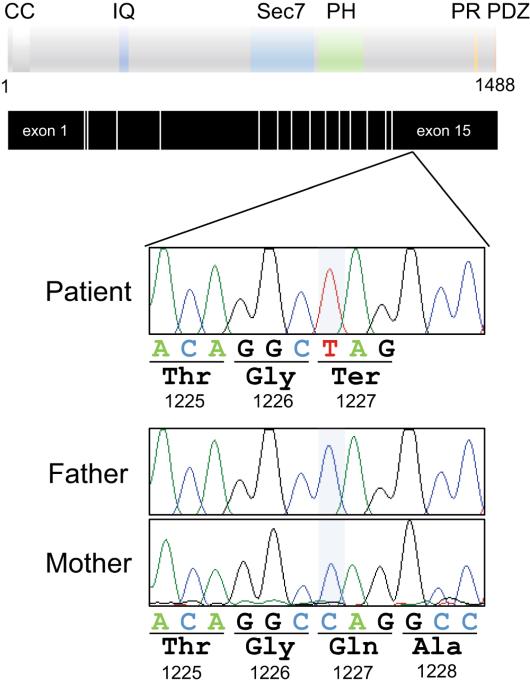
IQSEC2 gene on chromosome Xq11.22 encodes a member of guanine nucleotide exchange factor (GEF) protein that is implicated in the activation of ADP-ribosylation factors (Arfs) at the postsynaptic density (PSD), and plays a crucial role in synaptic transmission and dendritic spine formation. Alterations in IQSEC2 have been linked to X-linked intellectual developmental disorders including epilepsy and behavioral abnormalities. Of interest, truncating variants at the C-terminus of IQSEC2 can cause severe phenotypes, akin to truncating variants located in other regions. Here, we present a 5-year-old boy with severe intellectual disability and progressive epilepsy. The individual carried a nonsense variant p.Q1227* in the last exon of the IQSEC2 gene that was supposed to escape nonsense-mediated mRNA decay, thereby leading to a translation of C-terminus truncated IQSEC2 protein with residual activity. The functional analyses showed that the GEF activity of IQSEC2 Q1227* was compromised, and that the IQSEC2 Q1227* lacked preferential synaptic localization due to the absence of functional domains for binding to scaffolding proteins in the PSD. The impaired GEF activity and disrupted synaptic localization of the mutant IQSEC2 protein could impact dendritic and spine development in neurons, potentially explaining the patient’s severe neurological manifestations. Our findings indicate that C-terminal truncations in IQSEC2, previously not well-characterized, may have significant pathogenic implications.
期刊介绍:
The Journal of Human Genetics is an international journal publishing articles on human genetics, including medical genetics and human genome analysis. It covers all aspects of human genetics, including molecular genetics, clinical genetics, behavioral genetics, immunogenetics, pharmacogenomics, population genetics, functional genomics, epigenetics, genetic counseling and gene therapy.
Articles on the following areas are especially welcome: genetic factors of monogenic and complex disorders, genome-wide association studies, genetic epidemiology, cancer genetics, personal genomics, genotype-phenotype relationships and genome diversity.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
