{"title":"The formation and stability of 3D and 2D materials","authors":"Mona Layegh, Peng Yan, Joseph W. Bennett","doi":"10.1016/j.pcrysgrow.2023.100615","DOIUrl":null,"url":null,"abstract":"<div><p>With the emergence and popularity of high-performance computers, advances in materials informatics, and improvements in computing architectures and algorithms, the application of modeling in the field of materials science has become increasingly common and affordable. The ability to compute has benefited materials discovery in the last decade alone with many breakthroughs: improved photovoltaics, new functional nanomaterials, more efficient rechargeable batteries, and tailorable catalytic surfaces to name a few. Among various computing tools, first-principles calculations based on density functional theory (DFT) have been widely applied to high throughput computational analysis to better understand the formation, properties, and stability of new and existing materials. The advantages of DFT methods are that they are inexpensive, fast, and are capable of capturing nuances at the atomistic scale. Since DFT calculations are performed at 0 K and in vacuum, thermodynamic corrections need to be taken into account to match real world operating conditions in the laboratory and during use. These thermodynamic corrections have been applied for over twenty years and provided valuable guidance to the analysis of surface structure, vacancy formation, and stability across varying gaseous environments. The combination of DFT with experimental corrections significantly expands its flexibility as it can be used to generate stability conditions for specific elements and multi-component solids in water. This literature review will provide a thorough survey of first-principles DFT calculations combined with thermodynamics, as well as their application and research in the design, predicted stability, and characterization of 2D materials, their surfaces, and interfacial surface reactivity. A particular emphasis will be placed on the behavior of 2D materials in aqueous environments, comparing their surface transformation thermodynamics via processes such as ion release and adsorption using the newly created DFT + Solvent Ion Model (DSIM).</p></div>","PeriodicalId":409,"journal":{"name":"Progress in Crystal Growth and Characterization of Materials","volume":"70 1","pages":"Article 100615"},"PeriodicalIF":1.9000,"publicationDate":"2024-02-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.sciencedirect.com/science/article/pii/S0960897423000220/pdfft?md5=a2570dc205df4ac36d7b97d5b756a4b1&pid=1-s2.0-S0960897423000220-main.pdf","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Progress in Crystal Growth and Characterization of Materials","FirstCategoryId":"88","ListUrlMain":"https://www.sciencedirect.com/science/article/pii/S0960897423000220","RegionNum":2,"RegionCategory":"材料科学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2024/1/11 0:00:00","PubModel":"Epub","JCR":"Q1","JCRName":"CRYSTALLOGRAPHY","Score":null,"Total":0}
引用次数: 0
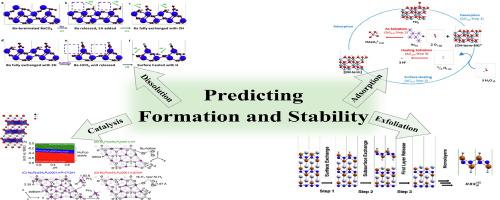
Abstract
With the emergence and popularity of high-performance computers, advances in materials informatics, and improvements in computing architectures and algorithms, the application of modeling in the field of materials science has become increasingly common and affordable. The ability to compute has benefited materials discovery in the last decade alone with many breakthroughs: improved photovoltaics, new functional nanomaterials, more efficient rechargeable batteries, and tailorable catalytic surfaces to name a few. Among various computing tools, first-principles calculations based on density functional theory (DFT) have been widely applied to high throughput computational analysis to better understand the formation, properties, and stability of new and existing materials. The advantages of DFT methods are that they are inexpensive, fast, and are capable of capturing nuances at the atomistic scale. Since DFT calculations are performed at 0 K and in vacuum, thermodynamic corrections need to be taken into account to match real world operating conditions in the laboratory and during use. These thermodynamic corrections have been applied for over twenty years and provided valuable guidance to the analysis of surface structure, vacancy formation, and stability across varying gaseous environments. The combination of DFT with experimental corrections significantly expands its flexibility as it can be used to generate stability conditions for specific elements and multi-component solids in water. This literature review will provide a thorough survey of first-principles DFT calculations combined with thermodynamics, as well as their application and research in the design, predicted stability, and characterization of 2D materials, their surfaces, and interfacial surface reactivity. A particular emphasis will be placed on the behavior of 2D materials in aqueous environments, comparing their surface transformation thermodynamics via processes such as ion release and adsorption using the newly created DFT + Solvent Ion Model (DSIM).
期刊介绍:
Materials especially crystalline materials provide the foundation of our modern technologically driven world. The domination of materials is achieved through detailed scientific research.
Advances in the techniques of growing and assessing ever more perfect crystals of a wide range of materials lie at the roots of much of today''s advanced technology. The evolution and development of crystalline materials involves research by dedicated scientists in academia as well as industry involving a broad field of disciplines including biology, chemistry, physics, material sciences and engineering. Crucially important applications in information technology, photonics, energy storage and harvesting, environmental protection, medicine and food production require a deep understanding of and control of crystal growth. This can involve suitable growth methods and material characterization from the bulk down to the nano-scale.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
