CRYAB stop-loss variant causes rare syndromic dilated cardiomyopathy with congenital cataract: expanding the phenotypic and mutational spectrum of alpha-B crystallinopathy
Changhee Ha, Darae Kim, Minjung Bak, Jong-Ho Park, Young-gon Kim, Ja-Hyun Jang, Jong-Won Kim, Jin-Oh Choi, Mi-Ae Jang
{"title":"CRYAB stop-loss variant causes rare syndromic dilated cardiomyopathy with congenital cataract: expanding the phenotypic and mutational spectrum of alpha-B crystallinopathy","authors":"Changhee Ha, Darae Kim, Minjung Bak, Jong-Ho Park, Young-gon Kim, Ja-Hyun Jang, Jong-Won Kim, Jin-Oh Choi, Mi-Ae Jang","doi":"10.1038/s10038-023-01218-1","DOIUrl":null,"url":null,"abstract":"Missense mutations in the alpha-B crystallin gene (CRYAB) have been reported in desmin-related myopathies with or without cardiomyopathy and have also been reported in families with only a cataract phenotype. Dilated cardiomyopathy (DCM) is a disorder with a highly heterogeneous genetic etiology involving more than 60 causative genes, hindering genetic diagnosis. In this study, we performed whole genome sequencing on 159 unrelated patients with DCM and identified an unusual stop-loss pathogenic variant in NM_001289808.2:c.527A>G of CRYAB in one patient. The mutant alpha-B crystallin protein is predicted to have an extended strand with addition of 19 amino acid residues, p.(Ter176TrpextTer19), which may contribute to aggregation and increased hydrophobicity of alpha-B crystallin. The proband, diagnosed with DCM at age 32, had a history of bilateral congenital cataracts but had no evidence of myopathy or associated symptoms. He also has a 10-year-old child diagnosed with bilateral congenital cataracts with the same CRYAB variant. This study expands the mutational spectrum of CRYAB and deepens our understanding of the complex phenotypes of alpha-B crystallinopathies.","PeriodicalId":16077,"journal":{"name":"Journal of Human Genetics","volume":"69 3-4","pages":"159-162"},"PeriodicalIF":2.5000,"publicationDate":"2024-01-12","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Human Genetics","FirstCategoryId":"99","ListUrlMain":"https://www.nature.com/articles/s10038-023-01218-1","RegionNum":3,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"GENETICS & HEREDITY","Score":null,"Total":0}
引用次数: 0
Abstract
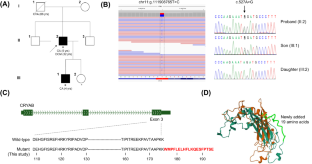
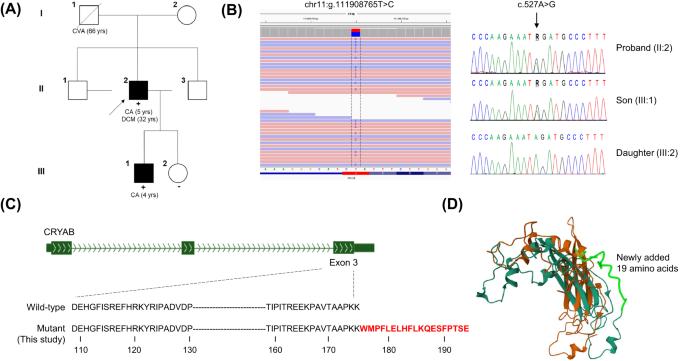
Missense mutations in the alpha-B crystallin gene (CRYAB) have been reported in desmin-related myopathies with or without cardiomyopathy and have also been reported in families with only a cataract phenotype. Dilated cardiomyopathy (DCM) is a disorder with a highly heterogeneous genetic etiology involving more than 60 causative genes, hindering genetic diagnosis. In this study, we performed whole genome sequencing on 159 unrelated patients with DCM and identified an unusual stop-loss pathogenic variant in NM_001289808.2:c.527A>G of CRYAB in one patient. The mutant alpha-B crystallin protein is predicted to have an extended strand with addition of 19 amino acid residues, p.(Ter176TrpextTer19), which may contribute to aggregation and increased hydrophobicity of alpha-B crystallin. The proband, diagnosed with DCM at age 32, had a history of bilateral congenital cataracts but had no evidence of myopathy or associated symptoms. He also has a 10-year-old child diagnosed with bilateral congenital cataracts with the same CRYAB variant. This study expands the mutational spectrum of CRYAB and deepens our understanding of the complex phenotypes of alpha-B crystallinopathies.
期刊介绍:
The Journal of Human Genetics is an international journal publishing articles on human genetics, including medical genetics and human genome analysis. It covers all aspects of human genetics, including molecular genetics, clinical genetics, behavioral genetics, immunogenetics, pharmacogenomics, population genetics, functional genomics, epigenetics, genetic counseling and gene therapy.
Articles on the following areas are especially welcome: genetic factors of monogenic and complex disorders, genome-wide association studies, genetic epidemiology, cancer genetics, personal genomics, genotype-phenotype relationships and genome diversity.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
