Identification of Two Variants c.2697A > C and c.3305A > C in USP7 by Analysis of Whole-Exome Sequencing in Chinese Patients with Hao-Fountain Syndrome.
Mei Sun, Qing Li, Ying Zhang, Yingzi Cai, Yan Dong, Jianbo Shu, Dong Li, Chunquan Cai
{"title":"Identification of Two Variants c.2697A > C and c.3305A > C in USP7 by Analysis of Whole-Exome Sequencing in Chinese Patients with Hao-Fountain Syndrome.","authors":"Mei Sun, Qing Li, Ying Zhang, Yingzi Cai, Yan Dong, Jianbo Shu, Dong Li, Chunquan Cai","doi":"10.1055/s-0043-1778089","DOIUrl":null,"url":null,"abstract":"<p><p><b>Background</b> Variants of ubiquitin-specific protease 7 ( <i>USP7</i> ) gene in humans are associated with a neurodevelopmental disorder-Hao-Fountain syndrome, its core symptoms including developmental delay, intellectual disability, and speech delay. Other variable symptoms can affect multiple systems. In present study, we report two patients with core features from two unrelated consanguineous families originating from the Tianjin Children's Hospital. <b>Methods and Results</b> Genomic DNA was extracted from the peripheral blood samples collected from the probands with their family members and whole-exome sequencing (WES) was used to detect the pathogenic genes in the probands. Suspected variants were subsequently validated by Sanger sequencing. In family 1, WES revealed that the proband carried the de novo variant c.2697A > C (p.Leu899Phe) in <i>USP7</i> (NM_003470.3). In family 2, WES identified the variant c.3305A > C (p.Asn1102Thr) in <i>USP7</i> (NM_003470.3) from the proband. <b>Conclusion</b> We reported two cases of Hao-Fountain syndrome caused by novel <i>USP7</i> variants. In addition, we report the first case of mosaicism with a <i>USP7</i> variant in Chinese family. Our findings demonstrate the importance of WES in diagnosis of genetic diseases and expands the <i>USP7</i> variants spectrum in Hao-Fountain syndrome. Moreover, we summarize the cases caused by <i>USP7</i> variants in the literature. Our study can provide a vital reference for the diagnosis of future cases.</p>","PeriodicalId":40142,"journal":{"name":"Global Medical Genetics","volume":"11 1","pages":"13-19"},"PeriodicalIF":1.5000,"publicationDate":"2024-01-16","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC10791489/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Global Medical Genetics","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1055/s-0043-1778089","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2024/1/1 0:00:00","PubModel":"eCollection","JCR":"Q4","JCRName":"GENETICS & HEREDITY","Score":null,"Total":0}
引用次数: 0
Abstract
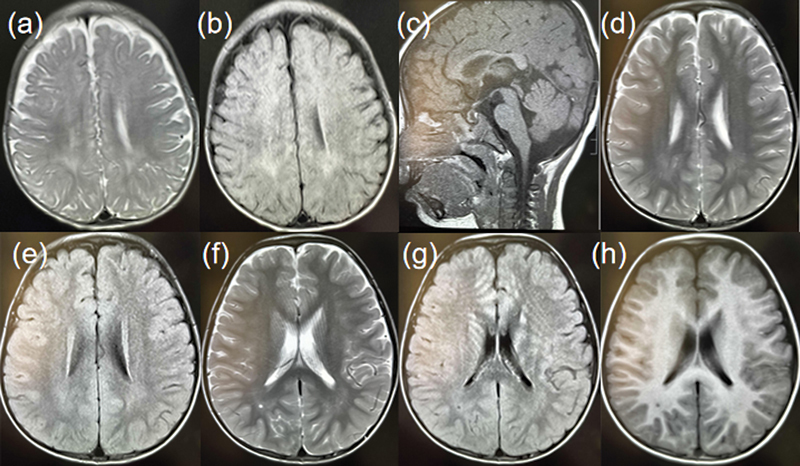
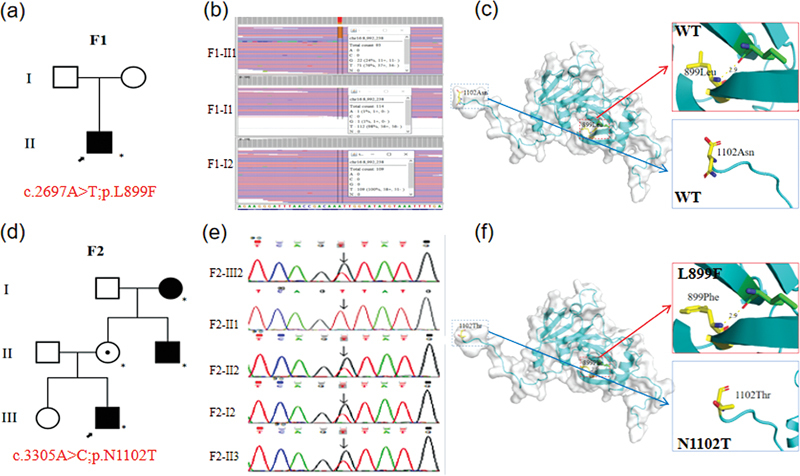
Background Variants of ubiquitin-specific protease 7 ( USP7 ) gene in humans are associated with a neurodevelopmental disorder-Hao-Fountain syndrome, its core symptoms including developmental delay, intellectual disability, and speech delay. Other variable symptoms can affect multiple systems. In present study, we report two patients with core features from two unrelated consanguineous families originating from the Tianjin Children's Hospital. Methods and Results Genomic DNA was extracted from the peripheral blood samples collected from the probands with their family members and whole-exome sequencing (WES) was used to detect the pathogenic genes in the probands. Suspected variants were subsequently validated by Sanger sequencing. In family 1, WES revealed that the proband carried the de novo variant c.2697A > C (p.Leu899Phe) in USP7 (NM_003470.3). In family 2, WES identified the variant c.3305A > C (p.Asn1102Thr) in USP7 (NM_003470.3) from the proband. Conclusion We reported two cases of Hao-Fountain syndrome caused by novel USP7 variants. In addition, we report the first case of mosaicism with a USP7 variant in Chinese family. Our findings demonstrate the importance of WES in diagnosis of genetic diseases and expands the USP7 variants spectrum in Hao-Fountain syndrome. Moreover, we summarize the cases caused by USP7 variants in the literature. Our study can provide a vital reference for the diagnosis of future cases.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
