Emilie Brûlé, Xiang Zhou, Ying Wang, Evan R S Buddle, Luisina Ongaro, Mary Loka, Anita Boelen, Daniel Bernard
{"title":"The hypothalamic-pituitary-thyroid axis is intact in male insulin receptor substrate 4 knockout mice.","authors":"Emilie Brûlé, Xiang Zhou, Ying Wang, Evan R S Buddle, Luisina Ongaro, Mary Loka, Anita Boelen, Daniel Bernard","doi":"10.1530/ETJ-23-0054","DOIUrl":null,"url":null,"abstract":"<p><strong>Objective: </strong>Loss of function mutations in the insulin receptor substrate 4 (IRS4) gene cause a rare form of X-linked congenital central hypothyroidism in boys and men. Affected individuals show decreased thyroid-stimulation hormone (TSH) secretion. Members of the IRS family canonically act as scaffold proteins between tyrosine kinase receptors and downstream effectors. How loss of IRS4 affects TSH synthesis or secretion is unresolved. We therefore assessed IRS4's role in the hypothalamic-pituitary-thyroid axis of Irs4 knockout mice.</p><p><strong>Methods: </strong>We generated two global Irs4 knockout mouse lines harboring either two or four base-pair deletions that result in frameshifts and loss of most of the IRS4 protein.</p><p><strong>Results: </strong>Under normal laboratory conditions, Irs4 knockout males did not exhibit impairments in pituitary expression of TSH subunit genes (Tshb or Cga) or in the thyrotropin-releasing hormone (TRH) receptor. Additionally, their serum thyroid hormone, T3 (triiodothyronine) and T4 (thyroxine), and hypothalamic Trh expression levels were normal. When Irs4 knockouts were rendered hypothyroid with a low-iodine diet supplemented with propylthiouracil (PTU) for 3 weeks, their serum TSH increased similarly to wild-type males.</p><p><strong>Conclusions: </strong>Overall, Irs4 knockout mice do not exhibit central hypothyroidism or otherwise appear to phenocopy IRS4 deficient patients. Compensation by another IRS protein may explain euthyroidism in these animals.</p>","PeriodicalId":12159,"journal":{"name":"European Thyroid Journal","volume":" ","pages":""},"PeriodicalIF":4.3000,"publicationDate":"2024-01-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC10895334/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"European Thyroid Journal","FirstCategoryId":"3","ListUrlMain":"https://doi.org/10.1530/ETJ-23-0054","RegionNum":2,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"ENDOCRINOLOGY & METABOLISM","Score":null,"Total":0}
引用次数: 0
Abstract
Objective: Loss of function mutations in the insulin receptor substrate 4 (IRS4) gene cause a rare form of X-linked congenital central hypothyroidism in boys and men. Affected individuals show decreased thyroid-stimulation hormone (TSH) secretion. Members of the IRS family canonically act as scaffold proteins between tyrosine kinase receptors and downstream effectors. How loss of IRS4 affects TSH synthesis or secretion is unresolved. We therefore assessed IRS4's role in the hypothalamic-pituitary-thyroid axis of Irs4 knockout mice.
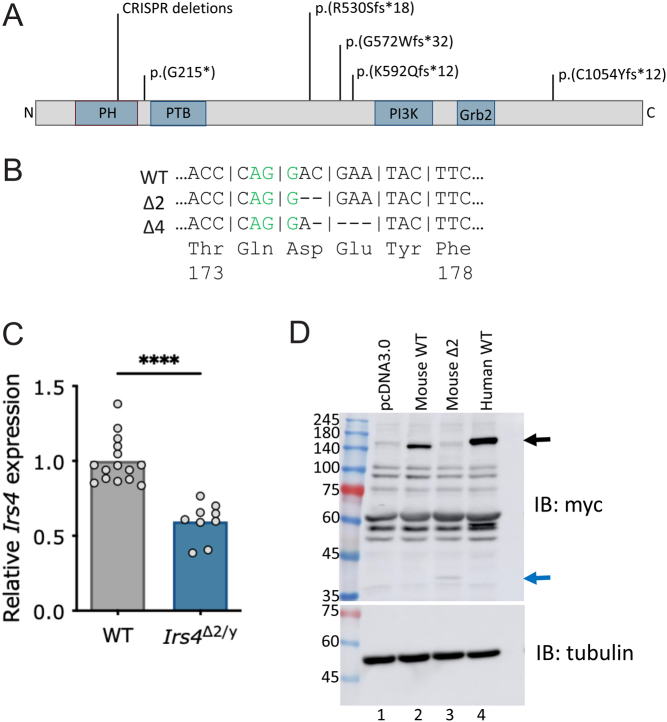
Methods: We generated two global Irs4 knockout mouse lines harboring either two or four base-pair deletions that result in frameshifts and loss of most of the IRS4 protein.
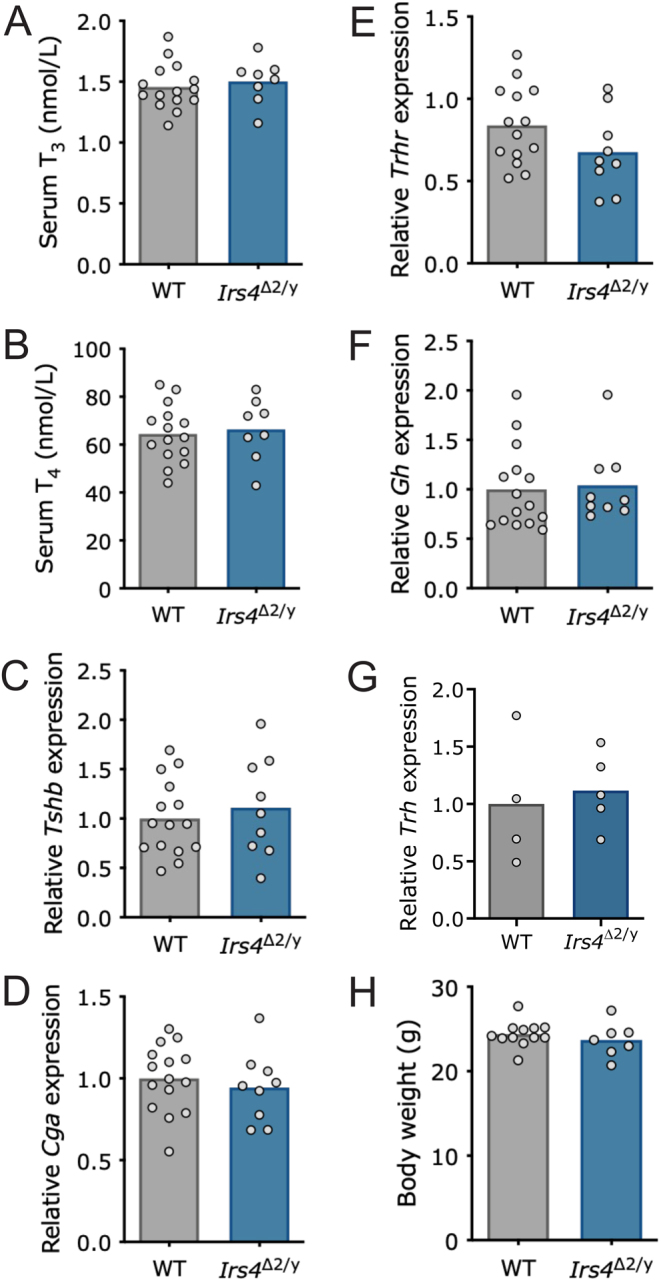
Results: Under normal laboratory conditions, Irs4 knockout males did not exhibit impairments in pituitary expression of TSH subunit genes (Tshb or Cga) or in the thyrotropin-releasing hormone (TRH) receptor. Additionally, their serum thyroid hormone, T3 (triiodothyronine) and T4 (thyroxine), and hypothalamic Trh expression levels were normal. When Irs4 knockouts were rendered hypothyroid with a low-iodine diet supplemented with propylthiouracil (PTU) for 3 weeks, their serum TSH increased similarly to wild-type males.
Conclusions: Overall, Irs4 knockout mice do not exhibit central hypothyroidism or otherwise appear to phenocopy IRS4 deficient patients. Compensation by another IRS protein may explain euthyroidism in these animals.
目的:胰岛素受体底物 4(IRS4)基因的功能缺失突变会导致男孩和男性患上一种罕见的 X 连锁先天性中枢性甲状腺功能减退症。患者的甲状腺刺激素(TSH)分泌减少。IRS 家族成员通常是酪氨酸激酶受体和下游效应器之间的支架蛋白。IRS4的缺失如何影响促甲状腺激素的合成或分泌,目前尚无定论。因此,我们评估了IRS4在Irs4基因敲除小鼠的下丘脑-垂体-甲状腺轴中的作用:方法:我们产生了两个Irs4基因全面敲除的小鼠品系,它们分别带有两个或四个碱基对的缺失,导致了IRS4蛋白的帧移位和大部分的缺失:结果:在正常实验室条件下,Irs4基因敲除的雄性小鼠并没有表现出垂体表达促甲状腺激素亚基基因(Tshb或Cga)或促甲状腺激素释放激素(TRH)受体的障碍。此外,它们的血清甲状腺激素、T3(三碘甲状腺原氨酸)和T4(甲状腺素)以及下丘脑Trh表达水平均正常。当用丙基硫脲嘧啶(PTU)补充低碘饮食使Irs4基因敲除者甲状腺功能减退3周时,它们的血清促甲状腺激素的增加与野生型雄性类似:总的来说,Irs4基因敲除小鼠不会表现出中枢性甲状腺功能减退症,也不会出现与IRS4缺乏症患者相似的表型。另一种IRS蛋白的补偿可能是这些动物甲状腺功能亢进的原因。
期刊介绍:
The ''European Thyroid Journal'' publishes papers reporting original research in basic, translational and clinical thyroidology. Original contributions cover all aspects of the field, from molecular and cellular biology to immunology and biochemistry, from physiology to pathology, and from pediatric to adult thyroid diseases with a special focus on thyroid cancer. Readers also benefit from reviews by noted experts, which highlight especially active areas of current research. The journal will further publish formal guidelines in the field, produced and endorsed by the European Thyroid Association.





 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
