Break Down of the Complexity and Inconsistency Between Levels of Matriglycan and Disease Phenotype in FKRP-Related Dystroglycanopathies: A Review and Model of Interpretation.
Qi L Lu, Molly C Holbrook, Marcela P Cataldi, Anthony Blaeser
{"title":"Break Down of the Complexity and Inconsistency Between Levels of Matriglycan and Disease Phenotype in FKRP-Related Dystroglycanopathies: A Review and Model of Interpretation.","authors":"Qi L Lu, Molly C Holbrook, Marcela P Cataldi, Anthony Blaeser","doi":"10.3233/JND-230205","DOIUrl":null,"url":null,"abstract":"<p><p>Dystroglycanopathies are a group of muscle degenerative diseases characterized with significant reduction in matriglycan expression critical in disease pathogenesis. Missense point mutations in the Fukutin-related protein (FKRP) gene cause variable reduction in the synthesis of matriglycan on alpha-dystroglycan (α-DG) and a wide range of disease severity. Data analyses of muscle biopsies from patients fail to show consistent correlation between the levels of matriglycan and clinical phenotypes. By reviewing clinical reports in conjunction with analysis of clinically relevant mouse models, we identify likely causes for the confusion. Nearly all missense FKRP mutations retain variable, but sufficient function for the synthesis of matriglycan during the later stage of muscle development and periods of muscle regeneration. These factors lead to a highly heterogenous pattern of matriglycan expression in diseased muscles, depending on age and stages of muscle regeneration. The limited size in clinical biopsy samples from different parts of even a single muscle tissue at different time points of disease progression may well mis-represent the residual function (base-levels) of the mutated FKRPs and phenotypes. We propose to use a simple Multi Point tool from ImageJ to more accurately measure the signal intensity of matriglycan expression on fiber membrane for assessing mutant FKRP function and therapeutic efficacy. A robust and sensitive immunohistochemical protocol would further improve reliability and comparability for the detection of matriglycan.</p>","PeriodicalId":16536,"journal":{"name":"Journal of neuromuscular diseases","volume":" ","pages":"275-284"},"PeriodicalIF":3.4000,"publicationDate":"2024-01-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC10977439/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of neuromuscular diseases","FirstCategoryId":"3","ListUrlMain":"https://doi.org/10.3233/JND-230205","RegionNum":4,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"CLINICAL NEUROLOGY","Score":null,"Total":0}
引用次数: 0
Abstract
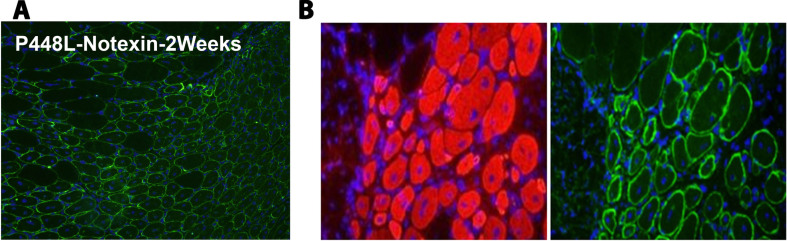
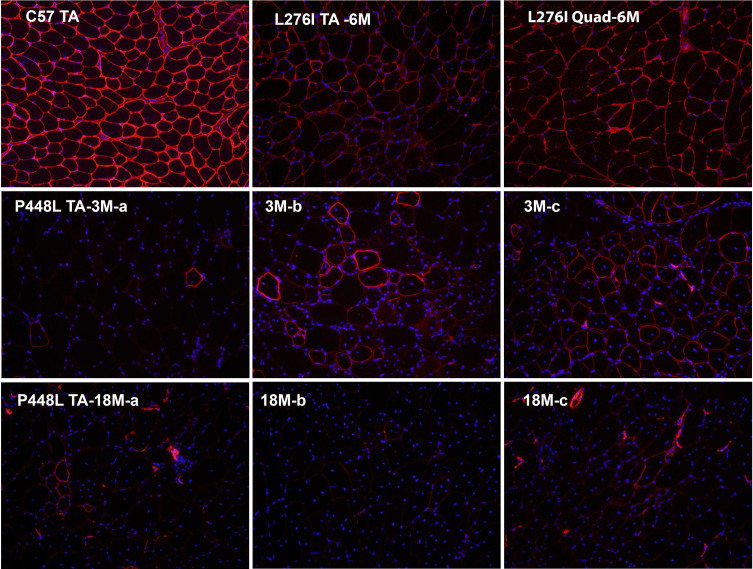
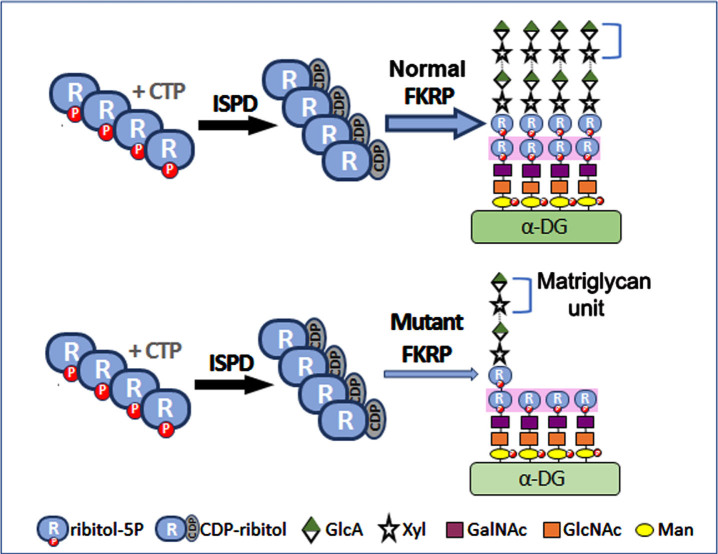
Dystroglycanopathies are a group of muscle degenerative diseases characterized with significant reduction in matriglycan expression critical in disease pathogenesis. Missense point mutations in the Fukutin-related protein (FKRP) gene cause variable reduction in the synthesis of matriglycan on alpha-dystroglycan (α-DG) and a wide range of disease severity. Data analyses of muscle biopsies from patients fail to show consistent correlation between the levels of matriglycan and clinical phenotypes. By reviewing clinical reports in conjunction with analysis of clinically relevant mouse models, we identify likely causes for the confusion. Nearly all missense FKRP mutations retain variable, but sufficient function for the synthesis of matriglycan during the later stage of muscle development and periods of muscle regeneration. These factors lead to a highly heterogenous pattern of matriglycan expression in diseased muscles, depending on age and stages of muscle regeneration. The limited size in clinical biopsy samples from different parts of even a single muscle tissue at different time points of disease progression may well mis-represent the residual function (base-levels) of the mutated FKRPs and phenotypes. We propose to use a simple Multi Point tool from ImageJ to more accurately measure the signal intensity of matriglycan expression on fiber membrane for assessing mutant FKRP function and therapeutic efficacy. A robust and sensitive immunohistochemical protocol would further improve reliability and comparability for the detection of matriglycan.
期刊介绍:
The Journal of Neuromuscular Diseases aims to facilitate progress in understanding the molecular genetics/correlates, pathogenesis, pharmacology, diagnosis and treatment of acquired and genetic neuromuscular diseases (including muscular dystrophy, myasthenia gravis, spinal muscular atrophy, neuropathies, myopathies, myotonias and myositis). The journal publishes research reports, reviews, short communications, letters-to-the-editor, and will consider research that has negative findings. The journal is dedicated to providing an open forum for original research in basic science, translational and clinical research that will improve our fundamental understanding and lead to effective treatments of neuromuscular diseases.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
