Sylvia Safwat, Kyle P Flannery, Ahmed A El Beheiry, Mohamed M Mokhtar, Ebtesam Abdalla, M Chiara Manzini
{"title":"Genetic blueprint of congenital muscular dystrophies with brain malformations in Egypt: A report of 11 families.","authors":"Sylvia Safwat, Kyle P Flannery, Ahmed A El Beheiry, Mohamed M Mokhtar, Ebtesam Abdalla, M Chiara Manzini","doi":"10.1007/s10048-024-00745-z","DOIUrl":null,"url":null,"abstract":"<p><p>Congenital muscular dystrophies (CMDs) are a group of rare muscle disorders characterized by early onset hypotonia and motor developmental delay associated with brain malformations with or without eye anomalies in the most severe cases. In this study, we aimed to uncover the genetic basis of severe CMD in Egypt and to determine the efficacy of whole exome sequencing (WES)-based genetic diagnosis in this population. We recruited twelve individuals from eleven families with a clinical diagnosis of CMD with brain malformations that fell into two groups: seven patients with suspected dystroglycanopathy and five patients with suspected merosin-deficient CMD. WES was analyzed by variant filtering using multiple approaches including splicing and copy number variant (CNV) analysis. We identified likely pathogenic variants in FKRP in two cases and variants in POMT1, POMK, and B3GALNT2 in three individuals. All individuals with merosin-deficient CMD had truncating variants in LAMA2. Further analysis in one of the two unsolved cases showed a homozygous protein-truncating variant in Feline Leukemia Virus subgroup C Receptor 1 (FLVCR1). FLVCR1 loss of function has never been previously reported. Yet, loss of function of its paralog, FLVCR2, causes lethal hydranencephaly-hydrocephaly syndrome (Fowler Syndrome) which should be considered in the differential diagnosis for dystroglycanopathy. Overall, we reached a diagnostic rate of 86% (6/7) for dystroglycanopathies and 100% (5/5) for merosinopathy. In conclusion, our results provide further evidence that WES is an important diagnostic method in CMD in developing countries to improve the diagnostic rate, management plan, and genetic counseling for these disorders.</p>","PeriodicalId":56106,"journal":{"name":"Neurogenetics","volume":" ","pages":"93-102"},"PeriodicalIF":1.2000,"publicationDate":"2024-04-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11076401/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Neurogenetics","FirstCategoryId":"3","ListUrlMain":"https://doi.org/10.1007/s10048-024-00745-z","RegionNum":4,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2024/2/1 0:00:00","PubModel":"Epub","JCR":"Q3","JCRName":"CLINICAL NEUROLOGY","Score":null,"Total":0}
引用次数: 0
Abstract
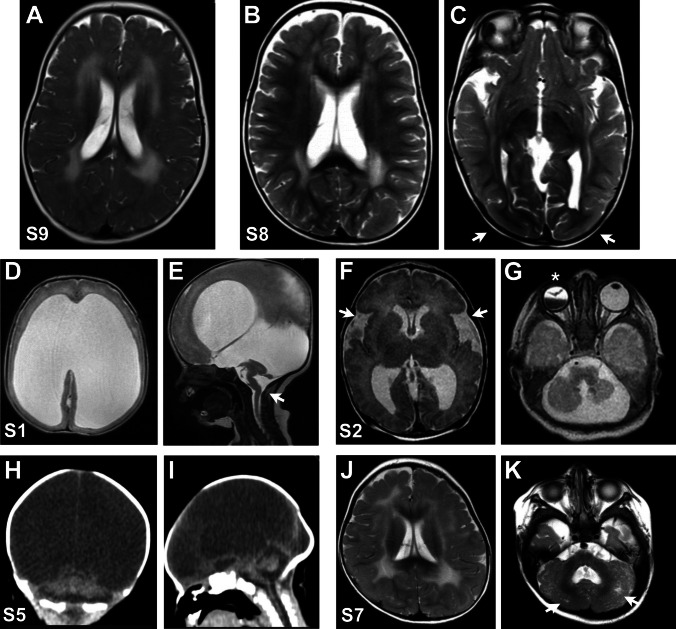
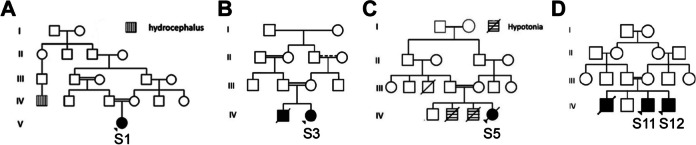
Congenital muscular dystrophies (CMDs) are a group of rare muscle disorders characterized by early onset hypotonia and motor developmental delay associated with brain malformations with or without eye anomalies in the most severe cases. In this study, we aimed to uncover the genetic basis of severe CMD in Egypt and to determine the efficacy of whole exome sequencing (WES)-based genetic diagnosis in this population. We recruited twelve individuals from eleven families with a clinical diagnosis of CMD with brain malformations that fell into two groups: seven patients with suspected dystroglycanopathy and five patients with suspected merosin-deficient CMD. WES was analyzed by variant filtering using multiple approaches including splicing and copy number variant (CNV) analysis. We identified likely pathogenic variants in FKRP in two cases and variants in POMT1, POMK, and B3GALNT2 in three individuals. All individuals with merosin-deficient CMD had truncating variants in LAMA2. Further analysis in one of the two unsolved cases showed a homozygous protein-truncating variant in Feline Leukemia Virus subgroup C Receptor 1 (FLVCR1). FLVCR1 loss of function has never been previously reported. Yet, loss of function of its paralog, FLVCR2, causes lethal hydranencephaly-hydrocephaly syndrome (Fowler Syndrome) which should be considered in the differential diagnosis for dystroglycanopathy. Overall, we reached a diagnostic rate of 86% (6/7) for dystroglycanopathies and 100% (5/5) for merosinopathy. In conclusion, our results provide further evidence that WES is an important diagnostic method in CMD in developing countries to improve the diagnostic rate, management plan, and genetic counseling for these disorders.
期刊介绍:
Neurogenetics publishes findings that contribute to a better understanding of the genetic basis of normal and abnormal function of the nervous system. Neurogenetic disorders are the main focus of the journal. Neurogenetics therefore includes findings in humans and other organisms that help understand neurological disease mechanisms and publishes papers from many different fields such as biophysics, cell biology, human genetics, neuroanatomy, neurochemistry, neurology, neuropathology, neurosurgery and psychiatry.
All papers submitted to Neurogenetics should be of sufficient immediate importance to justify urgent publication. They should present new scientific results. Data merely confirming previously published findings are not acceptable.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
