Case Presentation: Functional Assessment of a CASR Variant Identified in a Patient with Hypercalcaemia Confirms Familial Hypocalciuric Hypercalcaemia in the Patient and a Sister Previously Misdiagnosed with Primary Hyperparathyroidism.
Bryan K Ward, Kirsten A Loffell, John P Walsh, Warwick D Howe, Suzanne J Brown, Scott G Wilson
{"title":"Case Presentation: Functional Assessment of a <i>CASR</i> Variant Identified in a Patient with Hypercalcaemia Confirms Familial Hypocalciuric Hypercalcaemia in the Patient and a Sister Previously Misdiagnosed with Primary Hyperparathyroidism.","authors":"Bryan K Ward, Kirsten A Loffell, John P Walsh, Warwick D Howe, Suzanne J Brown, Scott G Wilson","doi":"10.1155/2024/6652801","DOIUrl":null,"url":null,"abstract":"<p><strong>Background: </strong>Primary hyperparathyroidism (PHPT) and familial hypocalciuric hypercalcaemia (FHH) are common causes of hypercalcaemia. Patients are mostly asymptomatic in the case of FHH and often so in the case of PHPT. In addition, biochemical parameters show considerable overlap, making differential diagnosis difficult. Genetic screening for inactivating variants in the calcium-sensing receptor (<i>CASR</i>) gene that are causative of FHH assists with the diagnosis since such variants are not generally associated with PHPT. However, novel <i>CASR</i> variants must undergo functional assessment before they can be definitively assigned a causative role in FHH. <i>Case Presentations</i>. We describe a 73-year-old female (patient A) who presented with mild parathyroid hormone (PTH)-dependent hypercalcaemia and a history of osteoporosis. Family history revealed that her sister (patient B) had presented a decade earlier with symptoms of PHPT including a history of mild hypercalcaemia and multiple renal calculi, prompting parathyroid surgery. However, a subtotal parathyroidectomy did not resolve her hypercalcaemia long term. On this basis, genetic screening was performed on patient A. This identified a heterozygous variant in the <i>CASR</i>, NM_000388.4:c.T101C: p.Leu34Pro (L34P). Functional analysis showed that the L34P variant was unable to produce mature, dimerized receptor and did not respond to Ca<sup>++</sup> ions. Adopting American College of Medical Genetics-based guidelines, the variant was classified as 'Pathogenic (II)'. Patient B was subsequently found to carry the L34P variant heterozygously, confirming a diagnosis of FHH, not PHPT.</p><p><strong>Conclusion: </strong>This study shows the importance of examining patient's family history in providing clues to the diagnosis in isolated cases of hypercalcaemia. In this case, history of a sister's unsuccessful parathyroidectomy prompted genetic screening in a patient who might otherwise have undergone inappropriate parathyroid surgery. Screening detected an inactivating <i>CASR</i> variant, firming up a diagnosis of FHH. These studies reaffirm the requirement for functionally assessing novel <i>CASR</i> variants prior to assigning causality to FHH.</p>","PeriodicalId":9621,"journal":{"name":"Case Reports in Endocrinology","volume":"2024 ","pages":"6652801"},"PeriodicalIF":0.9000,"publicationDate":"2024-02-03","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC10858793/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Case Reports in Endocrinology","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1155/2024/6652801","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2024/1/1 0:00:00","PubModel":"eCollection","JCR":"Q4","JCRName":"ENDOCRINOLOGY & METABOLISM","Score":null,"Total":0}
引用次数: 0
Abstract
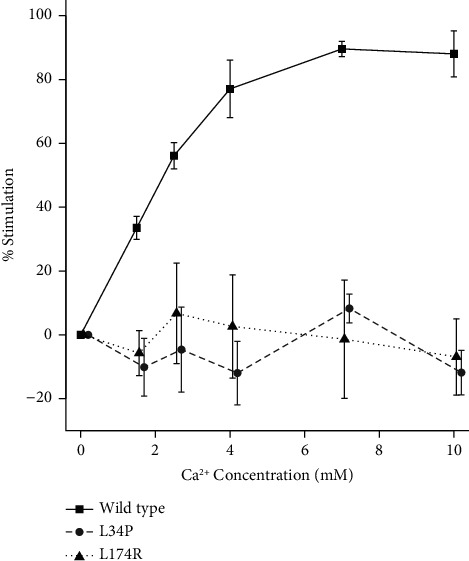
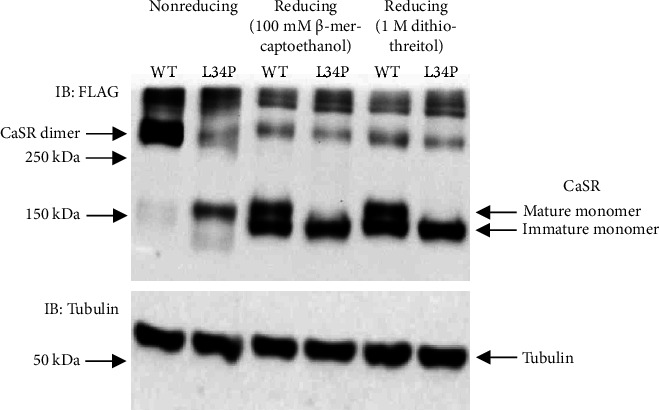
Background: Primary hyperparathyroidism (PHPT) and familial hypocalciuric hypercalcaemia (FHH) are common causes of hypercalcaemia. Patients are mostly asymptomatic in the case of FHH and often so in the case of PHPT. In addition, biochemical parameters show considerable overlap, making differential diagnosis difficult. Genetic screening for inactivating variants in the calcium-sensing receptor (CASR) gene that are causative of FHH assists with the diagnosis since such variants are not generally associated with PHPT. However, novel CASR variants must undergo functional assessment before they can be definitively assigned a causative role in FHH. Case Presentations. We describe a 73-year-old female (patient A) who presented with mild parathyroid hormone (PTH)-dependent hypercalcaemia and a history of osteoporosis. Family history revealed that her sister (patient B) had presented a decade earlier with symptoms of PHPT including a history of mild hypercalcaemia and multiple renal calculi, prompting parathyroid surgery. However, a subtotal parathyroidectomy did not resolve her hypercalcaemia long term. On this basis, genetic screening was performed on patient A. This identified a heterozygous variant in the CASR, NM_000388.4:c.T101C: p.Leu34Pro (L34P). Functional analysis showed that the L34P variant was unable to produce mature, dimerized receptor and did not respond to Ca++ ions. Adopting American College of Medical Genetics-based guidelines, the variant was classified as 'Pathogenic (II)'. Patient B was subsequently found to carry the L34P variant heterozygously, confirming a diagnosis of FHH, not PHPT.
Conclusion: This study shows the importance of examining patient's family history in providing clues to the diagnosis in isolated cases of hypercalcaemia. In this case, history of a sister's unsuccessful parathyroidectomy prompted genetic screening in a patient who might otherwise have undergone inappropriate parathyroid surgery. Screening detected an inactivating CASR variant, firming up a diagnosis of FHH. These studies reaffirm the requirement for functionally assessing novel CASR variants prior to assigning causality to FHH.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
