{"title":"A novel 2.4-kb PHKA2 deletion in a boy with glycogen storage disease type IXa","authors":"Takeshi Sato, Yosuke Ichihashi, Hideo Sugie, Tomohiro Ishii, Tomonobu Hasegawa","doi":"10.1111/cga.12555","DOIUrl":null,"url":null,"abstract":"<p>Glycogen storage disease type IXa (GSD IXa) results from a defect in the alpha subunit of phosphorylase kinase encoded by <i>PHKA2</i> with X-linked inheritance. Clinical manifestations include fasting hypoglycemia, hepatomegaly, and growth failure. Various <i>PHKA2</i> pathogenic variants have been reported, including microdeletions.<span><sup>1</sup></span> To date, few breakpoints have been identified, and mechanisms causing microdeletions are not well understood. We report on a pediatric patient with GSD IXa and a novel microdeletion in <i>PHKA2</i>.</p><p>The male proband was the first child of healthy, non-consanguineous Japanese parents. His birth length and weight were 48.4 cm (−0.29 SD) and 2.81 kg (−0.42 SD), respectively. Short stature was noted at 15 months of age; at 18 months, he could not stand alone and motor delay was suspected. He was referred to our hospital at 23 months with length and weight of 79.0 cm (−2.2 SD) and 10.5 kg (−0.82 SD), respectively. He could walk with aid and speak meaning words. Physical examination showed a doll-like appearance, abdominal distension, and hepatomegaly. Laboratory examinations showed as follows: white blood cells, 8800/μL (neutrophils 1584/μL); aspartate aminotransferase, 899 U/L; alanine aminotransferase, 554 U/L; creatine kinase 60 U/L; alkaline phosphatase 966 U/L (age matched reference, 395–1339); γ-glutamyl transpeptidase 374 U/L (reference, 6.5–60.0); creatinine, 0.13 mg/dL; uric acid, 7.0 mg/dL. Fasting glucagon loading test showed glucose of 42 mg/dL (before loading) and 64 mg/dL (60 min after loading). Two-hour postprandial glucagon loading test showed glucose of 131 mg/dL (before loading) and 191 mg/dL (30 min after loading). Oral glucose tolerance tests showed basal lactate and pyruvate levels of 7.0 and 0.61 mg/dL, respectively, and peak lactate and pyruvate levels of 32.2 mg/dL (90 min after loading) and 3.03 mg/dL (30 min after loading), respectively. Computed tomography showed hepatomegaly with high density. Phosphorylase kinase enzyme analysis of red blood cells revealed 0.3 nmoL/min/gHb, compared to 9.1 and 10.1 nmoL/min/gHb in two healthy subjects. We thus diagnosed him as having GSD IXa. After obtaining informed consent from his parents, genomic DNA was extracted from peripheral blood samples of the proband and his mother. We tried to amplify all exons and the flanking introns of the exons in <i>PHKA2</i> (NM_000292.3) in the proband, but we obtained no polymerase chain reaction (PCR) products of exons 20 and 21. We designed several new forward primers located at the intron intervening exons 19 and 20 and the reverse primer located at exon 22. Using these primers, we performed PCR using DNA from the proband or his mother and obtained the products, with the size at 500–650 bp from the proband and 3000 bp and 500–650 bp from his mother (Figure 1A). The PCR products in the proband were subjected to direct sequencing from both directions on the autosequencer. We identified a 2423 base deletion in <i>PHKA2</i> [NC_000023.11(NM_000292.3):c.2226+9_2360+360del] encompassing 134 nucleotides of exon 21 and a breakpoint with imperfect 8- to 9-bp and perfect 3- to 4-bp similar nucleotide sequences near the junction (Figure 1B). This 2.4-kb deletion has not been reported previously in patients with GSD IXa and was not found in the following databases: gnomAD SVs v4 (https://gnomad.broadinstitute.org/), 8.3KJPN-SV, and JSV1 databases (https://jmorp.megabank.tohoku.ac.jp/); however, one study reported a patient with the deletion of exon 21 in <i>PHKA2</i> detected by multiple ligation-dependent probe amplification.<span><sup>2</sup></span> No other pathogenic alterations were identified. Complementary DNA analysis in the proband revealed the skipping of exon 21, indicating that the deletion introduced a premature stop codon, p.Pro789Serfs*21 (Figure 1C). We speculate that this variant would trigger nonsense-mediated mRNA decay, based on a previous study using hepatocyte-like cells generated from the dermal fibroblast cell line of a male patient with <i>PHKA2</i> c.2597+5G>T, showing (i) the variant resulted in an aberrant splicing of <i>PHKA2</i> and the incorporation of a 27 bp into exon 23 with the immediate presence of a stop codon, (ii) <i>PHKA2</i> mRNA was down-regulated 7- to 11-fold, and (iii) mutant <i>PHKA2</i> protein expression was absent.<span><sup>3</sup></span></p><p>A previous study suggested that Alu-mediated recombination causes a 10-kb deletion of several exons in <i>PHKA2</i>.<span><sup>4</sup></span> To our knowledge, other mechanisms causing <i>PHKA2</i> microdeletions are unknown. In some diseases, microhomology at the junctions is considered to cause microdeletions through DNA rearrangement.<span><sup>5, 6</sup></span> Notably, in Duchenne and Becker muscular dystrophies, exon deletions within <i>DMD</i> occur possibly via microhomology of 2–5 bp at the junctions.<span><sup>6</sup></span> We speculate that microhomology, “gta” or “agta,” may have played an important role in DNA rearrangement in our patient. Meanwhile, the presence of imperfect direct repeats at mtDNA deletion breakpoints raised the possibility that our patient's imperfect 8- to 9-bp sequence may also contribute to the pathogenicity.<span><sup>7, 8</sup></span></p><p>In summary, we identified a novel 2.4-kb deletion in the <i>PHKA2</i> gene in a patient with GSD IXa. This case implies that DNA rearrangement via perfect and/or imperfect sequences may cause <i>PHKA2</i> microdeletion.</p><p>This study was supported by Novo Nordisk Pharma Ltd. and JCR Pharmaceuticals Co., Ltd.</p><p>The authors declare no conflicts of interest.</p><p>This study has been approved by the ethical committee at Keio University School of Medicine (20170130). We obtained written informed consent from legal guardians.</p>","PeriodicalId":10626,"journal":{"name":"Congenital Anomalies","volume":"64 2","pages":"63-65"},"PeriodicalIF":1.6000,"publicationDate":"2024-02-15","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://onlinelibrary.wiley.com/doi/epdf/10.1111/cga.12555","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Congenital Anomalies","FirstCategoryId":"3","ListUrlMain":"https://onlinelibrary.wiley.com/doi/10.1111/cga.12555","RegionNum":4,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q3","JCRName":"PEDIATRICS","Score":null,"Total":0}
引用次数: 0
Abstract
Glycogen storage disease type IXa (GSD IXa) results from a defect in the alpha subunit of phosphorylase kinase encoded by PHKA2 with X-linked inheritance. Clinical manifestations include fasting hypoglycemia, hepatomegaly, and growth failure. Various PHKA2 pathogenic variants have been reported, including microdeletions.1 To date, few breakpoints have been identified, and mechanisms causing microdeletions are not well understood. We report on a pediatric patient with GSD IXa and a novel microdeletion in PHKA2.
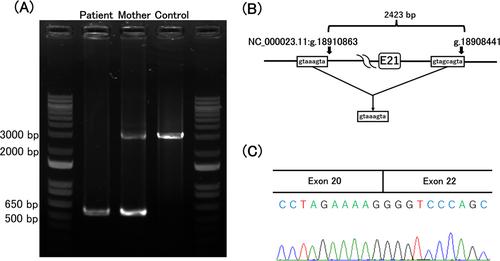
The male proband was the first child of healthy, non-consanguineous Japanese parents. His birth length and weight were 48.4 cm (−0.29 SD) and 2.81 kg (−0.42 SD), respectively. Short stature was noted at 15 months of age; at 18 months, he could not stand alone and motor delay was suspected. He was referred to our hospital at 23 months with length and weight of 79.0 cm (−2.2 SD) and 10.5 kg (−0.82 SD), respectively. He could walk with aid and speak meaning words. Physical examination showed a doll-like appearance, abdominal distension, and hepatomegaly. Laboratory examinations showed as follows: white blood cells, 8800/μL (neutrophils 1584/μL); aspartate aminotransferase, 899 U/L; alanine aminotransferase, 554 U/L; creatine kinase 60 U/L; alkaline phosphatase 966 U/L (age matched reference, 395–1339); γ-glutamyl transpeptidase 374 U/L (reference, 6.5–60.0); creatinine, 0.13 mg/dL; uric acid, 7.0 mg/dL. Fasting glucagon loading test showed glucose of 42 mg/dL (before loading) and 64 mg/dL (60 min after loading). Two-hour postprandial glucagon loading test showed glucose of 131 mg/dL (before loading) and 191 mg/dL (30 min after loading). Oral glucose tolerance tests showed basal lactate and pyruvate levels of 7.0 and 0.61 mg/dL, respectively, and peak lactate and pyruvate levels of 32.2 mg/dL (90 min after loading) and 3.03 mg/dL (30 min after loading), respectively. Computed tomography showed hepatomegaly with high density. Phosphorylase kinase enzyme analysis of red blood cells revealed 0.3 nmoL/min/gHb, compared to 9.1 and 10.1 nmoL/min/gHb in two healthy subjects. We thus diagnosed him as having GSD IXa. After obtaining informed consent from his parents, genomic DNA was extracted from peripheral blood samples of the proband and his mother. We tried to amplify all exons and the flanking introns of the exons in PHKA2 (NM_000292.3) in the proband, but we obtained no polymerase chain reaction (PCR) products of exons 20 and 21. We designed several new forward primers located at the intron intervening exons 19 and 20 and the reverse primer located at exon 22. Using these primers, we performed PCR using DNA from the proband or his mother and obtained the products, with the size at 500–650 bp from the proband and 3000 bp and 500–650 bp from his mother (Figure 1A). The PCR products in the proband were subjected to direct sequencing from both directions on the autosequencer. We identified a 2423 base deletion in PHKA2 [NC_000023.11(NM_000292.3):c.2226+9_2360+360del] encompassing 134 nucleotides of exon 21 and a breakpoint with imperfect 8- to 9-bp and perfect 3- to 4-bp similar nucleotide sequences near the junction (Figure 1B). This 2.4-kb deletion has not been reported previously in patients with GSD IXa and was not found in the following databases: gnomAD SVs v4 (https://gnomad.broadinstitute.org/), 8.3KJPN-SV, and JSV1 databases (https://jmorp.megabank.tohoku.ac.jp/); however, one study reported a patient with the deletion of exon 21 in PHKA2 detected by multiple ligation-dependent probe amplification.2 No other pathogenic alterations were identified. Complementary DNA analysis in the proband revealed the skipping of exon 21, indicating that the deletion introduced a premature stop codon, p.Pro789Serfs*21 (Figure 1C). We speculate that this variant would trigger nonsense-mediated mRNA decay, based on a previous study using hepatocyte-like cells generated from the dermal fibroblast cell line of a male patient with PHKA2 c.2597+5G>T, showing (i) the variant resulted in an aberrant splicing of PHKA2 and the incorporation of a 27 bp into exon 23 with the immediate presence of a stop codon, (ii) PHKA2 mRNA was down-regulated 7- to 11-fold, and (iii) mutant PHKA2 protein expression was absent.3
A previous study suggested that Alu-mediated recombination causes a 10-kb deletion of several exons in PHKA2.4 To our knowledge, other mechanisms causing PHKA2 microdeletions are unknown. In some diseases, microhomology at the junctions is considered to cause microdeletions through DNA rearrangement.5, 6 Notably, in Duchenne and Becker muscular dystrophies, exon deletions within DMD occur possibly via microhomology of 2–5 bp at the junctions.6 We speculate that microhomology, “gta” or “agta,” may have played an important role in DNA rearrangement in our patient. Meanwhile, the presence of imperfect direct repeats at mtDNA deletion breakpoints raised the possibility that our patient's imperfect 8- to 9-bp sequence may also contribute to the pathogenicity.7, 8
In summary, we identified a novel 2.4-kb deletion in the PHKA2 gene in a patient with GSD IXa. This case implies that DNA rearrangement via perfect and/or imperfect sequences may cause PHKA2 microdeletion.
This study was supported by Novo Nordisk Pharma Ltd. and JCR Pharmaceuticals Co., Ltd.
The authors declare no conflicts of interest.
This study has been approved by the ethical committee at Keio University School of Medicine (20170130). We obtained written informed consent from legal guardians.
期刊介绍:
Congenital Anomalies is the official English language journal of the Japanese Teratology Society, and publishes original articles in laboratory as well as clinical research in all areas of abnormal development and related fields, from all over the world. Although contributions by members of the teratology societies affiliated with The International Federation of Teratology Societies are given priority, contributions from non-members are welcomed.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
