Theoretical analysis of mechanism and regio- and stereoselectivity of 1, 3-dipolar cycloaddition of cyclic nitrone and substituted alkenes by DFT method
{"title":"Theoretical analysis of mechanism and regio- and stereoselectivity of 1, 3-dipolar cycloaddition of cyclic nitrone and substituted alkenes by DFT method","authors":"Samir Bouacha","doi":"10.1007/s11224-024-02292-7","DOIUrl":null,"url":null,"abstract":"<div><p>This study presents a theoretical investigation of the [3 + 2] cycloaddition (32CA) reaction between cyclic nitrone <b>a1</b> and substituted alkene <b>b1</b>. The mechanism, regioselectivity, and stereoselectivity of this 32CA reaction were analyzed using transition state theory and reactivity indices obtained from conceptual density functional theory (DFT) at the B3LYP/6-311G(d) level of theory. The results indicate that this cycloaddition reaction proceeds via an asynchronous one-step mechanism, exhibiting a non-polar nature and significant activation energies. These theoretical results are in agreement with the experimental observations. The study also employs topological analyses such as ESP, RDG-NCI, and ELF to determine active sites; distinguish hydrogen bonds, van der Waals interactions, and steric repulsive interactions; and predict electron localization, respectively.</p></div>","PeriodicalId":780,"journal":{"name":"Structural Chemistry","volume":"35 5","pages":"1427 - 1435"},"PeriodicalIF":2.2000,"publicationDate":"2024-02-14","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://link.springer.com/content/pdf/10.1007/s11224-024-02292-7.pdf","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Structural Chemistry","FirstCategoryId":"92","ListUrlMain":"https://link.springer.com/article/10.1007/s11224-024-02292-7","RegionNum":4,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q3","JCRName":"CHEMISTRY, MULTIDISCIPLINARY","Score":null,"Total":0}
引用次数: 0
Abstract
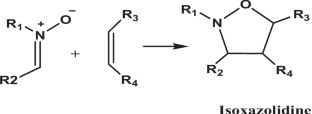
This study presents a theoretical investigation of the [3 + 2] cycloaddition (32CA) reaction between cyclic nitrone a1 and substituted alkene b1. The mechanism, regioselectivity, and stereoselectivity of this 32CA reaction were analyzed using transition state theory and reactivity indices obtained from conceptual density functional theory (DFT) at the B3LYP/6-311G(d) level of theory. The results indicate that this cycloaddition reaction proceeds via an asynchronous one-step mechanism, exhibiting a non-polar nature and significant activation energies. These theoretical results are in agreement with the experimental observations. The study also employs topological analyses such as ESP, RDG-NCI, and ELF to determine active sites; distinguish hydrogen bonds, van der Waals interactions, and steric repulsive interactions; and predict electron localization, respectively.
期刊介绍:
Structural Chemistry is an international forum for the publication of peer-reviewed original research papers that cover the condensed and gaseous states of matter and involve numerous techniques for the determination of structure and energetics, their results, and the conclusions derived from these studies. The journal overcomes the unnatural separation in the current literature among the areas of structure determination, energetics, and applications, as well as builds a bridge to other chemical disciplines. Ist comprehensive coverage encompasses broad discussion of results, observation of relationships among various properties, and the description and application of structure and energy information in all domains of chemistry.
We welcome the broadest range of accounts of research in structural chemistry involving the discussion of methodologies and structures,experimental, theoretical, and computational, and their combinations. We encourage discussions of structural information collected for their chemicaland biological significance.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
