{"title":"Bottom-Up Atomistic Descriptions of Top-Down Macroscopic Measurements: Computational Benchmarks for Hammett Electronic Parameters","authors":"Guilian Luchini, and , Robert S. Paton*, ","doi":"10.1021/acsphyschemau.3c00045","DOIUrl":null,"url":null,"abstract":"<p >The ability to relate substituent electronic effects to chemical reactivity is a cornerstone of physical organic chemistry and Linear Free Energy Relationships. The computation of electronic parameters is increasingly attractive since they can be obtained rapidly for structures and substituents without available experimental data and can be applied beyond aromatic substituents, for example, in studies of transition metal complexes and aliphatic and radical systems. Nevertheless, the description of “top-down” macroscopic observables, such as Hammett parameters using a “bottom-up” computational approach, poses several challenges for the practitioner. We have examined and benchmarked the performance of various computational charge schemes encompassing quantum mechanical methods that partition charge density, methods that fit charge to physical observables, and methods enhanced by semiempirical adjustments alongside NMR values. We study the locations of the atoms used to obtain these descriptors and their correlation with empirical Hammett parameters and rate differences resulting from electronic effects. These seemingly small choices have a much more significant impact than previously imagined, which outweighs the level of theory or basis set used. We observe a wide range of performance across the different computational protocols and observe stark and surprising differences in the ability of computational parameters to capture para- vs meta-electronic effects. In general, σ<sub>m</sub> predictions fare much worse than σ<sub>p</sub>. As a result, the choice of where to compute these descriptors─for the ring carbons or the attached H or other substituent atoms─affects their ability to capture experimental electronic differences. Density-based schemes, such as Hirshfeld charges, are more stable toward unphysical charge perturbations that result from nearby functional groups and outperform all other computational descriptors, including several commonly used basis set based schemes such as Natural Population Analysis. Using attached atoms also improves the statistical correlations. We obtained general linear relationships for the global prediction of experimental Hammett parameters from computed descriptors for use in statistical modeling studies.</p>","PeriodicalId":29796,"journal":{"name":"ACS Physical Chemistry Au","volume":"4 3","pages":"259–267"},"PeriodicalIF":4.3000,"publicationDate":"2024-02-06","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://pubs.acs.org/doi/epdf/10.1021/acsphyschemau.3c00045","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"ACS Physical Chemistry Au","FirstCategoryId":"1085","ListUrlMain":"https://pubs.acs.org/doi/10.1021/acsphyschemau.3c00045","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"CHEMISTRY, PHYSICAL","Score":null,"Total":0}
引用次数: 0
Abstract
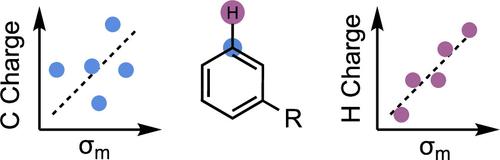
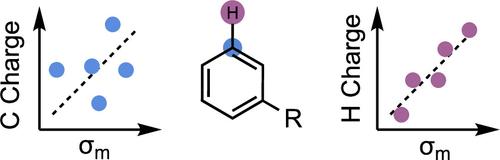
The ability to relate substituent electronic effects to chemical reactivity is a cornerstone of physical organic chemistry and Linear Free Energy Relationships. The computation of electronic parameters is increasingly attractive since they can be obtained rapidly for structures and substituents without available experimental data and can be applied beyond aromatic substituents, for example, in studies of transition metal complexes and aliphatic and radical systems. Nevertheless, the description of “top-down” macroscopic observables, such as Hammett parameters using a “bottom-up” computational approach, poses several challenges for the practitioner. We have examined and benchmarked the performance of various computational charge schemes encompassing quantum mechanical methods that partition charge density, methods that fit charge to physical observables, and methods enhanced by semiempirical adjustments alongside NMR values. We study the locations of the atoms used to obtain these descriptors and their correlation with empirical Hammett parameters and rate differences resulting from electronic effects. These seemingly small choices have a much more significant impact than previously imagined, which outweighs the level of theory or basis set used. We observe a wide range of performance across the different computational protocols and observe stark and surprising differences in the ability of computational parameters to capture para- vs meta-electronic effects. In general, σm predictions fare much worse than σp. As a result, the choice of where to compute these descriptors─for the ring carbons or the attached H or other substituent atoms─affects their ability to capture experimental electronic differences. Density-based schemes, such as Hirshfeld charges, are more stable toward unphysical charge perturbations that result from nearby functional groups and outperform all other computational descriptors, including several commonly used basis set based schemes such as Natural Population Analysis. Using attached atoms also improves the statistical correlations. We obtained general linear relationships for the global prediction of experimental Hammett parameters from computed descriptors for use in statistical modeling studies.
期刊介绍:
ACS Physical Chemistry Au is an open access journal which publishes original fundamental and applied research on all aspects of physical chemistry. The journal publishes new and original experimental computational and theoretical research of interest to physical chemists biophysical chemists chemical physicists physicists material scientists and engineers. An essential criterion for acceptance is that the manuscript provides new physical insight or develops new tools and methods of general interest. Some major topical areas include:Molecules Clusters and Aerosols; Biophysics Biomaterials Liquids and Soft Matter; Energy Materials and Catalysis


 分享
分享

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
