We present the cases of two sisters, both harboring the same ALDH18A1 gene mutations, who presented with a complex clinical phenotype characterized by spastic paraparesis with ataxia, epileptic encephalopathy, severe psychomotor deficits, and behavioral abnormalities.
Case description of two sisters with ALDH18A1 gene mutations.
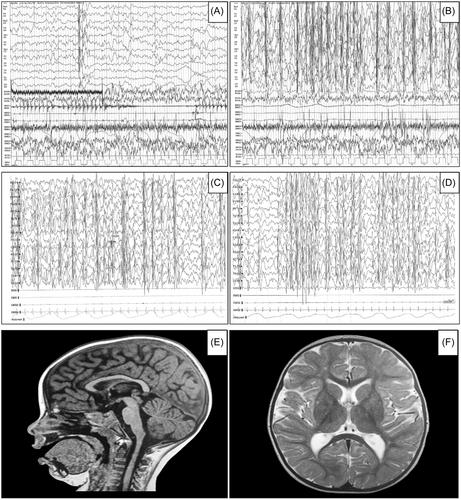
The older patient, a 12-year-old girl, exhibited spastic paraparesis with ataxia, microcephaly, facial dysmorphisms, and severe intellectual disability, with an absence of verbal language. An electroencephalogram (EEG) revealed marked spike-and-wave activation during sleep (SWAS), although no clinically documented seizures were observed. The younger sister, who was 9 years old, displayed a similar clinical presentation, including spastic paraparesis with ataxia, microcephaly, dysmorphisms, however, she displayed slightly more severe intellectual deficits and polymorphic seizures. EEG revealed a SWAS pattern in this case. Magnetic resonance imaging scans in both cases showed only a thin corpus callosum. Whole exome sequencing unveiled the presence of two likely pathogenic variants in compound heterozygosity within the ALDH18A1 gene. Specifically, these variants included the splice site variant c.88 + 1c.88+1G>A of paternal origin and the variant c.1364c.1364T>C (p.Leu455Ser) of maternal origin. Both sisters displayed normal blood levels of ammonia, ornithine, citrulline, arginine, and other amino acids.
These findings were compatible with ALDH18A1-related HSP complicated with a clinical and EEG pattern reminiscent of DEE-SWAS. We present the first report of DEE-SWAS in ALDH18A1-related HSP, expanding the clinical manifestations of this complex neurodevelopmental condition.


 分享
分享

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
