Ema Chaloupecká , Václav Tyrpekl , Kateřina Bártová , Yusuke Nishiyama , Martin Dračínský
{"title":"NMR crystallography of amino acids","authors":"Ema Chaloupecká , Václav Tyrpekl , Kateřina Bártová , Yusuke Nishiyama , Martin Dračínský","doi":"10.1016/j.ssnmr.2024.101921","DOIUrl":null,"url":null,"abstract":"<div><p>The development of NMR crystallography methods requires a reliable database of chemical shifts measured for systems with known crystal structure. We measured and assigned carbon and hydrogen chemical shifts of twenty solid natural amino acids of known polymorphic structure, meticulously determined using powder X-ray diffraction. We then correlated the experimental data with DFT-calculated isotropic shieldings. The small size of the unit cell of most amino acids allowed for advanced computations using various families of DFT functionals, including generalized gradient approximation (GGA), <em>meta</em>-GGA and hybrid DFT functionals. We tested several combinations of functionals for geometry optimizations and NMR calculations. For carbon shieldings, the widely used GGA functional PBE performed very well, although an improvement could be achieved by adding shielding corrections calculated for isolated molecules using a hybrid functional. For hydrogen nuclei, we observed the best performance for NMR calculations carried out with structures optimized at the hybrid DFT level. The high fidelity of the calculations made it possible to assign additional signals that could not be assigned based on experiments alone, for example signals of two non-equivalent molecules in the unit cell of some of the amino acids.</p></div>","PeriodicalId":21937,"journal":{"name":"Solid state nuclear magnetic resonance","volume":"130 ","pages":"Article 101921"},"PeriodicalIF":2.4000,"publicationDate":"2024-04-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Solid state nuclear magnetic resonance","FirstCategoryId":"92","ListUrlMain":"https://www.sciencedirect.com/science/article/pii/S0926204024000079","RegionNum":3,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2024/2/19 0:00:00","PubModel":"Epub","JCR":"Q4","JCRName":"CHEMISTRY, PHYSICAL","Score":null,"Total":0}
引用次数: 0
Abstract
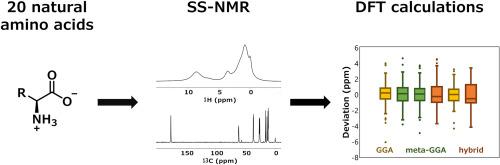
The development of NMR crystallography methods requires a reliable database of chemical shifts measured for systems with known crystal structure. We measured and assigned carbon and hydrogen chemical shifts of twenty solid natural amino acids of known polymorphic structure, meticulously determined using powder X-ray diffraction. We then correlated the experimental data with DFT-calculated isotropic shieldings. The small size of the unit cell of most amino acids allowed for advanced computations using various families of DFT functionals, including generalized gradient approximation (GGA), meta-GGA and hybrid DFT functionals. We tested several combinations of functionals for geometry optimizations and NMR calculations. For carbon shieldings, the widely used GGA functional PBE performed very well, although an improvement could be achieved by adding shielding corrections calculated for isolated molecules using a hybrid functional. For hydrogen nuclei, we observed the best performance for NMR calculations carried out with structures optimized at the hybrid DFT level. The high fidelity of the calculations made it possible to assign additional signals that could not be assigned based on experiments alone, for example signals of two non-equivalent molecules in the unit cell of some of the amino acids.
期刊介绍:
The journal Solid State Nuclear Magnetic Resonance publishes original manuscripts of high scientific quality dealing with all experimental and theoretical aspects of solid state NMR. This includes advances in instrumentation, development of new experimental techniques and methodology, new theoretical insights, new data processing and simulation methods, and original applications of established or novel methods to scientific problems.



 分享
分享

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
