Sebastian Ciro Acosta, Lorena Díaz-Ordóñez, Juan David Gutierrez-Medina, Yisther Katherine Silva-Cuero, Luis Guillermo Arango-Vélez, Andrés Octavio García-Trujillo, Harry Pachajoa
{"title":"Familial LCAT Deficiency and Low HDL-C Levels: In silico Characterization of Two Rare LCAT Missense Mutations.","authors":"Sebastian Ciro Acosta, Lorena Díaz-Ordóñez, Juan David Gutierrez-Medina, Yisther Katherine Silva-Cuero, Luis Guillermo Arango-Vélez, Andrés Octavio García-Trujillo, Harry Pachajoa","doi":"10.2147/TACG.S438135","DOIUrl":null,"url":null,"abstract":"<p><p>Mutations in the lecithin-cholesterol acyltransferase (<i>LCAT</i>) gene, which catalyzes the esterification of cholesterol, result in two types of autosomal recessive disorders: Familial <i>LCAT</i> deficiency (FLD) and Fish Eye Disease (FED). While both phenotypes are characterized by corneal opacities and different forms of dyslipidemia, such as low levels of high-density lipoprotein-cholesterol (HDL-C), FLD exhibits more severe clinical manifestations like splenomegaly, anemia, and renal failure. We describe the first clinically and genetically confirmed case of FLD in Colombia which corresponds to a 46-year-old woman with corneal opacity, hypothyroidism, and dyslipidemia, who does not have any manifestations of renal failure, with two pathogenic heterozygous missense variants in the <i>LCAT</i> gene: <i>LCAT</i> (NM_000229.2):c.803G>A (p.Arg268His) and <i>LCAT</i> (NM_000229.2):c.368G>C (p.Arg123Pro). In silico analysis of the mutations predicted the physicochemical properties of the mutated protein, causing instability and potentially decreased <i>LCAT</i> function. These compound mutations highlight the clinical heterogeneity of the phenotypes associated with <i>LCAT</i> gene mutations.</p>","PeriodicalId":39131,"journal":{"name":"Application of Clinical Genetics","volume":"17 ","pages":"23-32"},"PeriodicalIF":2.6000,"publicationDate":"2024-02-20","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC10893891/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Application of Clinical Genetics","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.2147/TACG.S438135","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2024/1/1 0:00:00","PubModel":"eCollection","JCR":"Q2","JCRName":"GENETICS & HEREDITY","Score":null,"Total":0}
引用次数: 0
Abstract
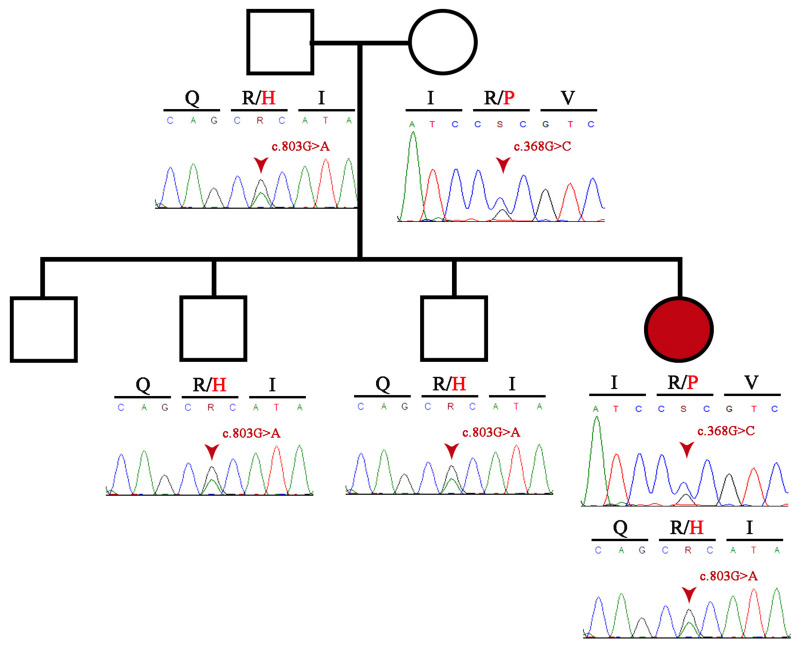
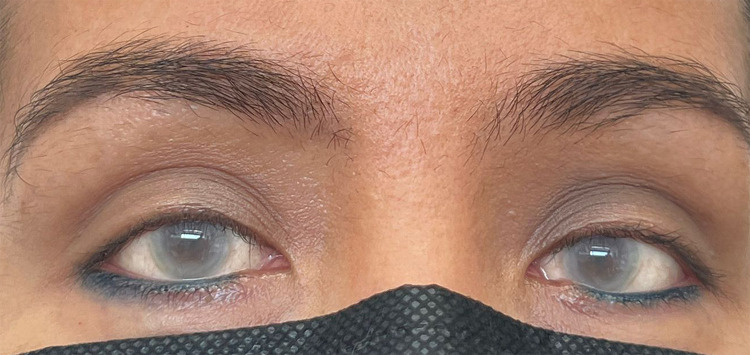
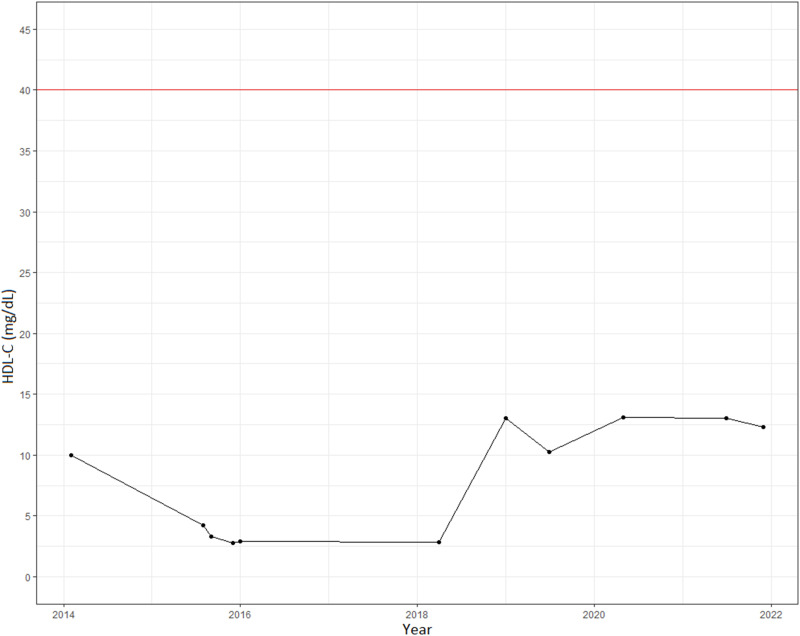
Mutations in the lecithin-cholesterol acyltransferase (LCAT) gene, which catalyzes the esterification of cholesterol, result in two types of autosomal recessive disorders: Familial LCAT deficiency (FLD) and Fish Eye Disease (FED). While both phenotypes are characterized by corneal opacities and different forms of dyslipidemia, such as low levels of high-density lipoprotein-cholesterol (HDL-C), FLD exhibits more severe clinical manifestations like splenomegaly, anemia, and renal failure. We describe the first clinically and genetically confirmed case of FLD in Colombia which corresponds to a 46-year-old woman with corneal opacity, hypothyroidism, and dyslipidemia, who does not have any manifestations of renal failure, with two pathogenic heterozygous missense variants in the LCAT gene: LCAT (NM_000229.2):c.803G>A (p.Arg268His) and LCAT (NM_000229.2):c.368G>C (p.Arg123Pro). In silico analysis of the mutations predicted the physicochemical properties of the mutated protein, causing instability and potentially decreased LCAT function. These compound mutations highlight the clinical heterogeneity of the phenotypes associated with LCAT gene mutations.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
