Sara Massucco, Chiara Gemelli, Emilia Bellone, Alessandro Geroldi, Serena Patrone, Paola Mandich, Elena Scarsi, Elena Faedo, Lucio Marinelli, Tiziana Mongini, Monica Traverso, Serena Baratto, Angelo Schenone, Chiara Fiorillo, Marina Grandis
{"title":"Skeletal muscle involvement in biallelic <i>SORD</i> mutations: case report and review of the literature.","authors":"Sara Massucco, Chiara Gemelli, Emilia Bellone, Alessandro Geroldi, Serena Patrone, Paola Mandich, Elena Scarsi, Elena Faedo, Lucio Marinelli, Tiziana Mongini, Monica Traverso, Serena Baratto, Angelo Schenone, Chiara Fiorillo, Marina Grandis","doi":"10.36185/2532-1900-323","DOIUrl":null,"url":null,"abstract":"<p><p>Biallelic mutations in the sorbitol dehydrogenase (<i>SORD</i>) gene have been identified as a genetic cause of autosomal recessive axonal Charcot-Marie-Tooth disease 2 (CMT2) and distal hereditary motor neuropathy (dHMN). We herein review the main phenotypes associated with <i>SORD</i> mutations and report the case of a 16-year-old man who was referred to our outpatient clinic for a slowly worsening gait disorder with wasting and weakness of distal lower limbs musculature. Since creatine phosphokinase (CPK) values were persistently raised (1.5fold increased) and a Next-Generation Sequencing CMT-associated panel failed in identifying pathogenic variants, a muscle biopsy was performed with evidence of alterations suggestive of a protein surplus distal myopathy. Finally, Whole-Exome Sequencing (WES) identified two pathogenic <i>SORD</i> variants in the heterozygous state: c.458C > A (p.Ala153Asp) and c.757delG (p.Ala253Glnfs*27). This is an isolated report of compound heterozygosity for two <i>SORD</i> mutations associated with clinical and histological signs of skeletal muscle involvement, expanding the phenotypic expression of <i>SORD</i> mutations.</p>","PeriodicalId":93851,"journal":{"name":"Acta myologica : myopathies and cardiomyopathies : official journal of the Mediterranean Society of Myology","volume":"42 4","pages":"113-117"},"PeriodicalIF":0.0000,"publicationDate":"2023-12-20","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC10883325/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Acta myologica : myopathies and cardiomyopathies : official journal of the Mediterranean Society of Myology","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.36185/2532-1900-323","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2023/1/1 0:00:00","PubModel":"eCollection","JCR":"","JCRName":"","Score":null,"Total":0}
引用次数: 0
Abstract
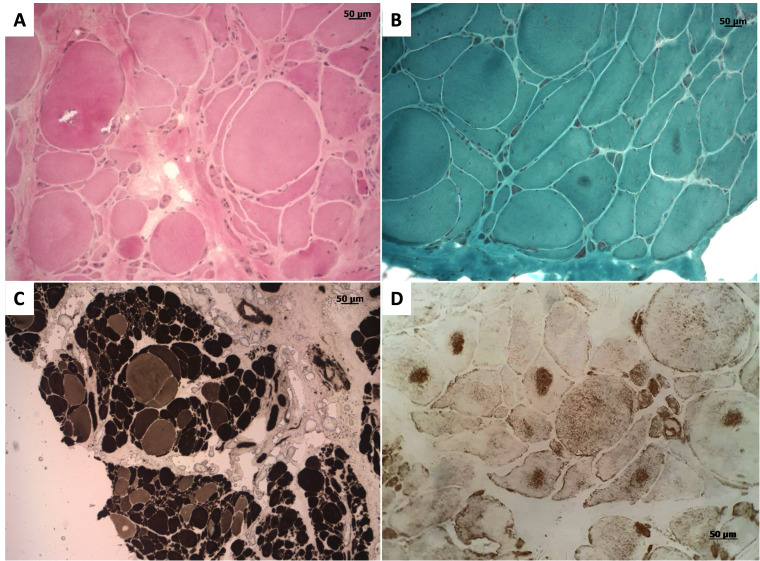
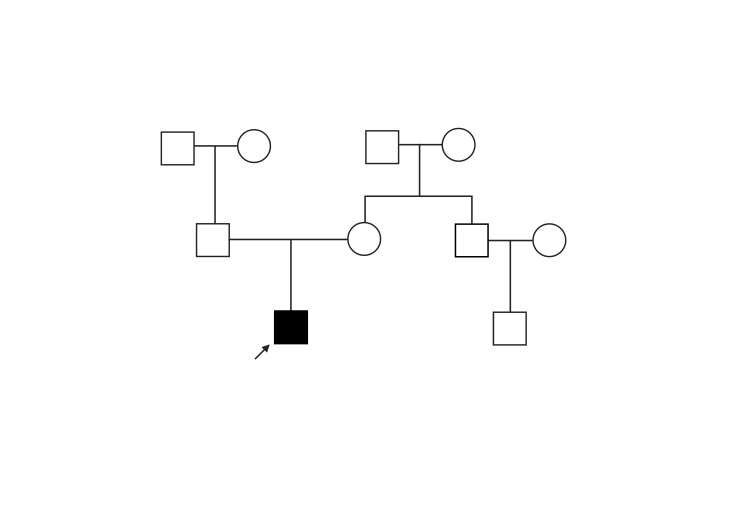
Biallelic mutations in the sorbitol dehydrogenase (SORD) gene have been identified as a genetic cause of autosomal recessive axonal Charcot-Marie-Tooth disease 2 (CMT2) and distal hereditary motor neuropathy (dHMN). We herein review the main phenotypes associated with SORD mutations and report the case of a 16-year-old man who was referred to our outpatient clinic for a slowly worsening gait disorder with wasting and weakness of distal lower limbs musculature. Since creatine phosphokinase (CPK) values were persistently raised (1.5fold increased) and a Next-Generation Sequencing CMT-associated panel failed in identifying pathogenic variants, a muscle biopsy was performed with evidence of alterations suggestive of a protein surplus distal myopathy. Finally, Whole-Exome Sequencing (WES) identified two pathogenic SORD variants in the heterozygous state: c.458C > A (p.Ala153Asp) and c.757delG (p.Ala253Glnfs*27). This is an isolated report of compound heterozygosity for two SORD mutations associated with clinical and histological signs of skeletal muscle involvement, expanding the phenotypic expression of SORD mutations.


 分享
分享

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
