Daniel Brooks, Elizabeth Burke, Sukyeong Lee, Tanya N Eble, Melanie O'Leary, Ikeoluwa Osei-Owusu, Heidi L Rehm, Shweta U Dhar, Lisa Emrick, David Bick, Michelle Nehrebecky, Ellen Macnamara, Dídac Casas-Alba, Judith Armstrong, Carolina Prat, Antonio F Martínez-Monseny, Francesc Palau, Pengfei Liu, David Adams, Seema Lalani, Jill A Rosenfeld, Lindsay C Burrage
{"title":"Heterozygous MAP3K20 variants cause ectodermal dysplasia, craniosynostosis, sensorineural hearing loss, and limb anomalies.","authors":"Daniel Brooks, Elizabeth Burke, Sukyeong Lee, Tanya N Eble, Melanie O'Leary, Ikeoluwa Osei-Owusu, Heidi L Rehm, Shweta U Dhar, Lisa Emrick, David Bick, Michelle Nehrebecky, Ellen Macnamara, Dídac Casas-Alba, Judith Armstrong, Carolina Prat, Antonio F Martínez-Monseny, Francesc Palau, Pengfei Liu, David Adams, Seema Lalani, Jill A Rosenfeld, Lindsay C Burrage","doi":"10.1007/s00439-024-02657-2","DOIUrl":null,"url":null,"abstract":"<p><p>Biallelic pathogenic variants in MAP3K20, which encodes a mitogen-activated protein kinase, are a rare cause of split-hand foot malformation (SHFM), hearing loss, and nail abnormalities or congenital myopathy. However, heterozygous variants in this gene have not been definitively associated with a phenotype. Here, we describe the phenotypic spectrum associated with heterozygous de novo variants in the linker region between the kinase domain and leucine zipper domain of MAP3K20. We report five individuals with diverse clinical features, including craniosynostosis, limb anomalies, sensorineural hearing loss, and ectodermal dysplasia-like phenotypes who have heterozygous de novo variants in this specific region of the gene. These individuals exhibit both shared and unique clinical manifestations, highlighting the complexity and variability of the disorder. We propose that the involvement of MAP3K20 in endothelial-mesenchymal transition provides a plausible etiology of these features. Together, these findings characterize a disorder that both expands the phenotypic spectrum associated with MAP3K20 and highlights the need for further studies on its role in early human development.</p>","PeriodicalId":13175,"journal":{"name":"Human Genetics","volume":" ","pages":"279-291"},"PeriodicalIF":3.6000,"publicationDate":"2024-03-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11191325/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Human Genetics","FirstCategoryId":"99","ListUrlMain":"https://doi.org/10.1007/s00439-024-02657-2","RegionNum":2,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2024/3/7 0:00:00","PubModel":"Epub","JCR":"Q2","JCRName":"GENETICS & HEREDITY","Score":null,"Total":0}
引用次数: 0
Abstract
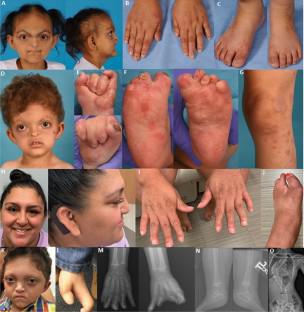
Biallelic pathogenic variants in MAP3K20, which encodes a mitogen-activated protein kinase, are a rare cause of split-hand foot malformation (SHFM), hearing loss, and nail abnormalities or congenital myopathy. However, heterozygous variants in this gene have not been definitively associated with a phenotype. Here, we describe the phenotypic spectrum associated with heterozygous de novo variants in the linker region between the kinase domain and leucine zipper domain of MAP3K20. We report five individuals with diverse clinical features, including craniosynostosis, limb anomalies, sensorineural hearing loss, and ectodermal dysplasia-like phenotypes who have heterozygous de novo variants in this specific region of the gene. These individuals exhibit both shared and unique clinical manifestations, highlighting the complexity and variability of the disorder. We propose that the involvement of MAP3K20 in endothelial-mesenchymal transition provides a plausible etiology of these features. Together, these findings characterize a disorder that both expands the phenotypic spectrum associated with MAP3K20 and highlights the need for further studies on its role in early human development.
期刊介绍:
Human Genetics is a monthly journal publishing original and timely articles on all aspects of human genetics. The Journal particularly welcomes articles in the areas of Behavioral genetics, Bioinformatics, Cancer genetics and genomics, Cytogenetics, Developmental genetics, Disease association studies, Dysmorphology, ELSI (ethical, legal and social issues), Evolutionary genetics, Gene expression, Gene structure and organization, Genetics of complex diseases and epistatic interactions, Genetic epidemiology, Genome biology, Genome structure and organization, Genotype-phenotype relationships, Human Genomics, Immunogenetics and genomics, Linkage analysis and genetic mapping, Methods in Statistical Genetics, Molecular diagnostics, Mutation detection and analysis, Neurogenetics, Physical mapping and Population Genetics. Articles reporting animal models relevant to human biology or disease are also welcome. Preference will be given to those articles which address clinically relevant questions or which provide new insights into human biology.
Unless reporting entirely novel and unusual aspects of a topic, clinical case reports, cytogenetic case reports, papers on descriptive population genetics, articles dealing with the frequency of polymorphisms or additional mutations within genes in which numerous lesions have already been described, and papers that report meta-analyses of previously published datasets will normally not be accepted.
The Journal typically will not consider for publication manuscripts that report merely the isolation, map position, structure, and tissue expression profile of a gene of unknown function unless the gene is of particular interest or is a candidate gene involved in a human trait or disorder.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
