Split hand/foot malformation (SHFM) is a congenital limb disorder presenting with limb anomalies, such as missing, hypoplastic, or fused digits, and often craniofacial defects, including a cleft lip/palate, microdontia, micrognathia, or maxillary hypoplasia. We previously identified three novel variants in the transcription factor, PRDM1, that are associated with SHFM phenotypes. One individual also presented with a high arch palate. Studies in vertebrates indicate that PRDM1 is important for development of the skull; however, prior to our study, human variants in PRDM1 had not been associated with craniofacial anomalies.
Using transient mRNA overexpression assays in prdm1a−/− mutant zebrafish, we tested whether the PRDM1 SHFM variants were functional and could lead to a rescue of the craniofacial defects observed in prdm1a−/− mutants. We also mined previously published CUT&RUN and RNA-seq datasets that sorted EGFP-positive cells from a Tg(Mmu:Prx1-EGFP) transgenic line that labels the pectoral fin, pharyngeal arches, and dorsal part of the head to examine Prdm1a binding and the effect of Prdm1a loss on craniofacial genes.
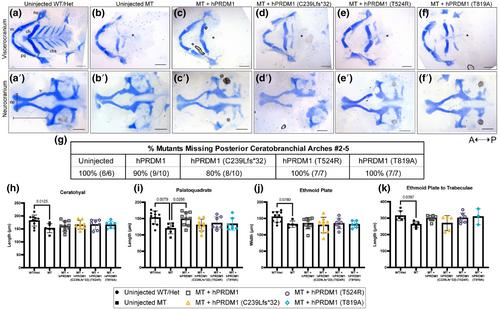
The prdm1a−/− mutants exhibit craniofacial defects including a hypoplastic neurocranium, a loss of posterior ceratobranchial arches, a shorter palatoquadrate, and an inverted ceratohyal. Injection of wildtype (WT) hPRDM1 in prdm1a−/− mutants partially rescues the palatoquadrate phenotype. However, injection of each of the three SHFM variants fails to rescue this skeletal defect. Loss of prdm1a leads to a decreased expression of important craniofacial genes by RNA-seq, including emilin3a, confirmed by hybridization chain reaction expression. Other genes including dlx5a/dlx6a, hand2, sox9b, col2a1a, and hoxb genes are also reduced. Validation by real-time quantitative PCR in the anterior half of zebrafish embryos failed to confirm the expression changes suggesting that the differences are enriched in prx1 expressing cells.
These data suggest that the three SHFM variants are likely not functional and may be associated with the craniofacial defects observed in the humans. Finally, they demonstrate how Prdm1a can directly bind and regulate genes involved in craniofacial development.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
