Yanlin Zhang, Christopher J F Cameron, Mathieu Blanchette
{"title":"Posterior inference of Hi-C contact frequency through sampling.","authors":"Yanlin Zhang, Christopher J F Cameron, Mathieu Blanchette","doi":"10.3389/fbinf.2023.1285828","DOIUrl":null,"url":null,"abstract":"<p><p>Hi-C is one of the most widely used approaches to study three-dimensional genome conformations. Contacts captured by a Hi-C experiment are represented in a contact frequency matrix. Due to the limited sequencing depth and other factors, Hi-C contact frequency matrices are only approximations of the true interaction frequencies and are further reported without any quantification of uncertainty. Hence, downstream analyses based on Hi-C contact maps (e.g., TAD and loop annotation) are themselves point estimations. Here, we present the Hi-C interaction frequency sampler (HiCSampler) that reliably infers the posterior distribution of the interaction frequency for a given Hi-C contact map by exploiting dependencies between neighboring loci. Posterior predictive checks demonstrate that HiCSampler can infer highly predictive chromosomal interaction frequency. Summary statistics calculated by HiCSampler provide a measurement of the uncertainty for Hi-C experiments, and samples inferred by HiCSampler are ready for use by most downstream analysis tools off the shelf and permit uncertainty measurements in these analyses without modifications.</p>","PeriodicalId":73066,"journal":{"name":"Frontiers in bioinformatics","volume":"3 ","pages":"1285828"},"PeriodicalIF":3.9000,"publicationDate":"2024-02-22","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC10919286/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Frontiers in bioinformatics","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.3389/fbinf.2023.1285828","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2023/1/1 0:00:00","PubModel":"eCollection","JCR":"Q2","JCRName":"MATHEMATICAL & COMPUTATIONAL BIOLOGY","Score":null,"Total":0}
引用次数: 0
Abstract
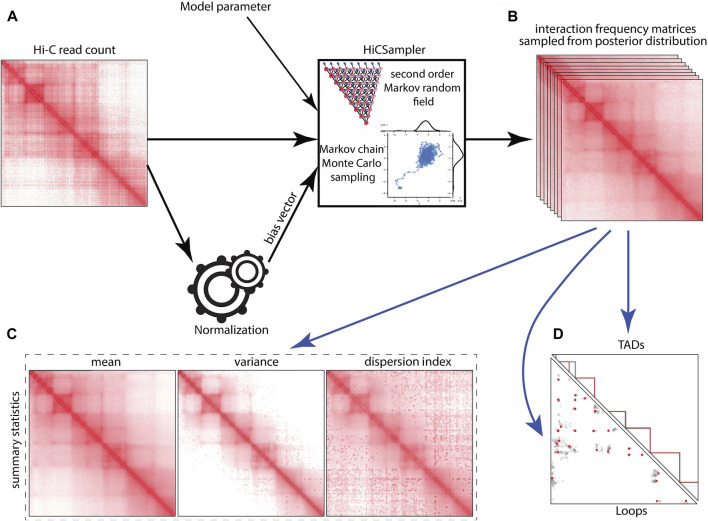
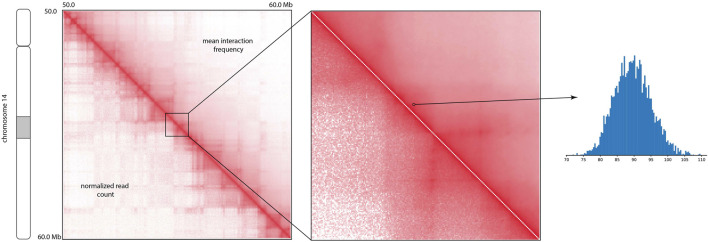
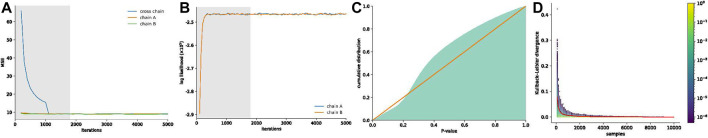
Hi-C is one of the most widely used approaches to study three-dimensional genome conformations. Contacts captured by a Hi-C experiment are represented in a contact frequency matrix. Due to the limited sequencing depth and other factors, Hi-C contact frequency matrices are only approximations of the true interaction frequencies and are further reported without any quantification of uncertainty. Hence, downstream analyses based on Hi-C contact maps (e.g., TAD and loop annotation) are themselves point estimations. Here, we present the Hi-C interaction frequency sampler (HiCSampler) that reliably infers the posterior distribution of the interaction frequency for a given Hi-C contact map by exploiting dependencies between neighboring loci. Posterior predictive checks demonstrate that HiCSampler can infer highly predictive chromosomal interaction frequency. Summary statistics calculated by HiCSampler provide a measurement of the uncertainty for Hi-C experiments, and samples inferred by HiCSampler are ready for use by most downstream analysis tools off the shelf and permit uncertainty measurements in these analyses without modifications.


 分享
分享

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
