{"title":"New insights into the clinical and molecular spectrum of the MADD-related neurodevelopmental disorder","authors":"Ghada M. H. Abdel-Salam, Mohamed S. Abdel-Hamid","doi":"10.1038/s10038-024-01236-7","DOIUrl":null,"url":null,"abstract":"Biallelic pathogenic variants in MADD lead to a very rare neurodevelopmental disorder which is phenotypically pleiotropic grossly ranging from severe neonatal hypotonia, failure to thrive, multiple organ dysfunction, and early lethality to a similar but milder phenotype with better survival. Here, we report 5 patients from 3 unrelated Egyptian families in whom 4 patients showed the severe end of the spectrum displaying neonatal respiratory distress, hypotonia and chronic diarrhea while one patient presented with the mild form displaying moderate intellectual disability and myopathy. In addition, we observed distal arthrogryposis and nonspecific structural brain anomalies in all our patients. Interestingly, cerebellar and brainstem hypoplasia were noted in one patient. Whole exome sequencing identified three novel homozygous variants in the MADD gene: two likely pathogenic [c.4321delC p.(Gln1441ArgfsTer46) and c.2620 C > T p.(Arg874Ter)] and one variant of uncertain significance (c.4307 G > A, p.Arg1436Gln). The variants segregated with the disease in all available family members. Our findings confirm that arthrogryposis, genital, cardiac and structural brain anomalies are manifestations of MADD which expand the spectrum of MADD-related neurodevelopmental disorder. Moreover, they further highlight the convergence of MADD variants on different organ systems leading to complex phenotypes.","PeriodicalId":16077,"journal":{"name":"Journal of Human Genetics","volume":"69 6","pages":"263-270"},"PeriodicalIF":2.5000,"publicationDate":"2024-03-08","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.nature.com/articles/s10038-024-01236-7.pdf","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Human Genetics","FirstCategoryId":"99","ListUrlMain":"https://www.nature.com/articles/s10038-024-01236-7","RegionNum":3,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"GENETICS & HEREDITY","Score":null,"Total":0}
引用次数: 0
Abstract
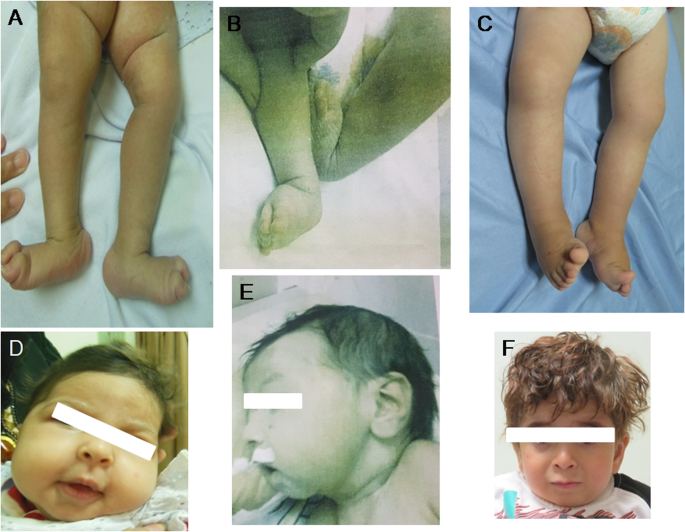
Biallelic pathogenic variants in MADD lead to a very rare neurodevelopmental disorder which is phenotypically pleiotropic grossly ranging from severe neonatal hypotonia, failure to thrive, multiple organ dysfunction, and early lethality to a similar but milder phenotype with better survival. Here, we report 5 patients from 3 unrelated Egyptian families in whom 4 patients showed the severe end of the spectrum displaying neonatal respiratory distress, hypotonia and chronic diarrhea while one patient presented with the mild form displaying moderate intellectual disability and myopathy. In addition, we observed distal arthrogryposis and nonspecific structural brain anomalies in all our patients. Interestingly, cerebellar and brainstem hypoplasia were noted in one patient. Whole exome sequencing identified three novel homozygous variants in the MADD gene: two likely pathogenic [c.4321delC p.(Gln1441ArgfsTer46) and c.2620 C > T p.(Arg874Ter)] and one variant of uncertain significance (c.4307 G > A, p.Arg1436Gln). The variants segregated with the disease in all available family members. Our findings confirm that arthrogryposis, genital, cardiac and structural brain anomalies are manifestations of MADD which expand the spectrum of MADD-related neurodevelopmental disorder. Moreover, they further highlight the convergence of MADD variants on different organ systems leading to complex phenotypes.
期刊介绍:
The Journal of Human Genetics is an international journal publishing articles on human genetics, including medical genetics and human genome analysis. It covers all aspects of human genetics, including molecular genetics, clinical genetics, behavioral genetics, immunogenetics, pharmacogenomics, population genetics, functional genomics, epigenetics, genetic counseling and gene therapy.
Articles on the following areas are especially welcome: genetic factors of monogenic and complex disorders, genome-wide association studies, genetic epidemiology, cancer genetics, personal genomics, genotype-phenotype relationships and genome diversity.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
