Expanding the genetic and phenotypic spectrum of TRAPPC9 and MID2-related neurodevelopmental disabilities: report of two novel mutations, 3D-modelling, and molecular docking studies
{"title":"Expanding the genetic and phenotypic spectrum of TRAPPC9 and MID2-related neurodevelopmental disabilities: report of two novel mutations, 3D-modelling, and molecular docking studies","authors":"Marwa Kharrat, Chahnez Triki, Abir ben isaa, Wafa Bouchaala, Olfa Alila, Jihen Chouchen, Yosra Ghouliya, Fatma Kamoun, Abdelaziz Tlili, Faiza Fakhfakh","doi":"10.1038/s10038-024-01242-9","DOIUrl":null,"url":null,"abstract":"Intellectual disabilities (ID) and autism spectrum disorders (ASD) have a variety of etiologies, including environmental and genetic factors. Our study reports a psychiatric clinical investigation and a molecular analysis using whole exome sequencing (WES) of two siblings with ID and ASD from a consanguineous family. Bioinformatic prediction and molecular docking analysis were also carried out. The two patients were diagnosed with profound intellectual disability, brain malformations such as cortical atrophy, acquired microcephaly, and autism level III. The neurological and neuropsychiatric examination revealed that P2 was more severely affected than P1, as he was unable to walk, presented with dysmorphic feature and exhibited self and hetero aggressive behaviors. The molecular investigations revealed a novel TRAPPC9 biallelic nonsense mutation (c.2920 C > T, p.R974X) in the two siblings. The more severely affected patient (P2) presented, along with the TRAPPC9 variant, a new missense mutation c.166 C > T (p.R56C) in the MID2 gene at hemizygous state, while his sister P1 was merely a carrier. The 3D modelling and molecular docking analysis revealed that c.166 C > T variant could affect the ability of MID2 binding to Astrin, leading to dysregulation of microtubule dynamics and causing morphological abnormalities in the brain. As our knowledge, the MID2 mutation (p.R56C) is the first one to be detected in Tunisia and causing phenotypic variability between the siblings. We extend the genetic and clinical spectrum of TRAPPC9 and MID2 mutations and highlights the possible concomitant presence of X-linked as well as autosomal recessive inheritance to causing ID, microcephaly, and autism.","PeriodicalId":16077,"journal":{"name":"Journal of Human Genetics","volume":"69 7","pages":"291-299"},"PeriodicalIF":2.5000,"publicationDate":"2024-03-11","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Human Genetics","FirstCategoryId":"99","ListUrlMain":"https://www.nature.com/articles/s10038-024-01242-9","RegionNum":3,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"GENETICS & HEREDITY","Score":null,"Total":0}
引用次数: 0
Abstract
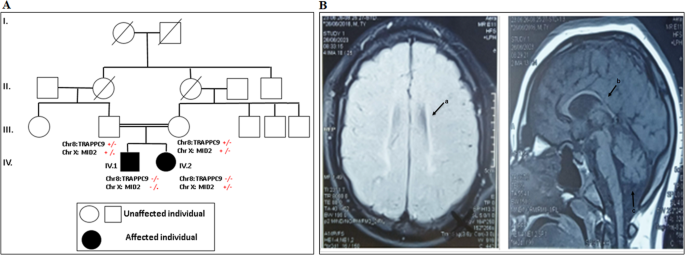
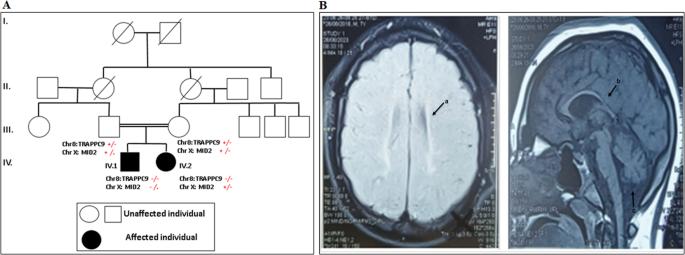
Intellectual disabilities (ID) and autism spectrum disorders (ASD) have a variety of etiologies, including environmental and genetic factors. Our study reports a psychiatric clinical investigation and a molecular analysis using whole exome sequencing (WES) of two siblings with ID and ASD from a consanguineous family. Bioinformatic prediction and molecular docking analysis were also carried out. The two patients were diagnosed with profound intellectual disability, brain malformations such as cortical atrophy, acquired microcephaly, and autism level III. The neurological and neuropsychiatric examination revealed that P2 was more severely affected than P1, as he was unable to walk, presented with dysmorphic feature and exhibited self and hetero aggressive behaviors. The molecular investigations revealed a novel TRAPPC9 biallelic nonsense mutation (c.2920 C > T, p.R974X) in the two siblings. The more severely affected patient (P2) presented, along with the TRAPPC9 variant, a new missense mutation c.166 C > T (p.R56C) in the MID2 gene at hemizygous state, while his sister P1 was merely a carrier. The 3D modelling and molecular docking analysis revealed that c.166 C > T variant could affect the ability of MID2 binding to Astrin, leading to dysregulation of microtubule dynamics and causing morphological abnormalities in the brain. As our knowledge, the MID2 mutation (p.R56C) is the first one to be detected in Tunisia and causing phenotypic variability between the siblings. We extend the genetic and clinical spectrum of TRAPPC9 and MID2 mutations and highlights the possible concomitant presence of X-linked as well as autosomal recessive inheritance to causing ID, microcephaly, and autism.
期刊介绍:
The Journal of Human Genetics is an international journal publishing articles on human genetics, including medical genetics and human genome analysis. It covers all aspects of human genetics, including molecular genetics, clinical genetics, behavioral genetics, immunogenetics, pharmacogenomics, population genetics, functional genomics, epigenetics, genetic counseling and gene therapy.
Articles on the following areas are especially welcome: genetic factors of monogenic and complex disorders, genome-wide association studies, genetic epidemiology, cancer genetics, personal genomics, genotype-phenotype relationships and genome diversity.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
