{"title":"Leveraging machine learning models for peptide–protein interaction prediction","authors":"Song Yin, Xuenan Mi and Diwakar Shukla","doi":"10.1039/D3CB00208J","DOIUrl":null,"url":null,"abstract":"<p >Peptides play a pivotal role in a wide range of biological activities through participating in up to 40% protein–protein interactions in cellular processes. They also demonstrate remarkable specificity and efficacy, making them promising candidates for drug development. However, predicting peptide–protein complexes by traditional computational approaches, such as docking and molecular dynamics simulations, still remains a challenge due to high computational cost, flexible nature of peptides, and limited structural information of peptide–protein complexes. In recent years, the surge of available biological data has given rise to the development of an increasing number of machine learning models for predicting peptide–protein interactions. These models offer efficient solutions to address the challenges associated with traditional computational approaches. Furthermore, they offer enhanced accuracy, robustness, and interpretability in their predictive outcomes. This review presents a comprehensive overview of machine learning and deep learning models that have emerged in recent years for the prediction of peptide–protein interactions.</p>","PeriodicalId":40691,"journal":{"name":"RSC Chemical Biology","volume":" 5","pages":" 401-417"},"PeriodicalIF":3.1000,"publicationDate":"2024-03-13","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://pubs.rsc.org/en/content/articlepdf/2024/cb/d3cb00208j?page=search","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"RSC Chemical Biology","FirstCategoryId":"1085","ListUrlMain":"https://pubs.rsc.org/en/content/articlelanding/2024/cb/d3cb00208j","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"BIOCHEMISTRY & MOLECULAR BIOLOGY","Score":null,"Total":0}
引用次数: 0
Abstract
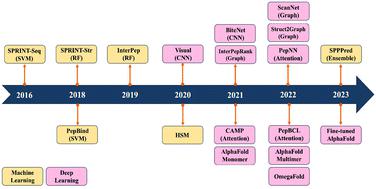
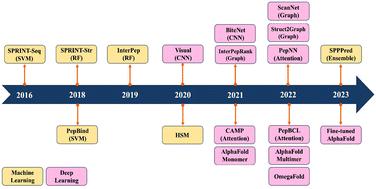
Peptides play a pivotal role in a wide range of biological activities through participating in up to 40% protein–protein interactions in cellular processes. They also demonstrate remarkable specificity and efficacy, making them promising candidates for drug development. However, predicting peptide–protein complexes by traditional computational approaches, such as docking and molecular dynamics simulations, still remains a challenge due to high computational cost, flexible nature of peptides, and limited structural information of peptide–protein complexes. In recent years, the surge of available biological data has given rise to the development of an increasing number of machine learning models for predicting peptide–protein interactions. These models offer efficient solutions to address the challenges associated with traditional computational approaches. Furthermore, they offer enhanced accuracy, robustness, and interpretability in their predictive outcomes. This review presents a comprehensive overview of machine learning and deep learning models that have emerged in recent years for the prediction of peptide–protein interactions.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
