Eva Klinman, Catherine Gooch, Joel S Perlmutter, Albert A Davis, Baijayanta Maiti
{"title":"Novel <i>SGCE</i> Mutation in a Patient With Myoclonus-Dystonia: A Case Report.","authors":"Eva Klinman, Catherine Gooch, Joel S Perlmutter, Albert A Davis, Baijayanta Maiti","doi":"10.1212/NXG.0000000000200128","DOIUrl":null,"url":null,"abstract":"<p><strong>Objectives: </strong>Characterize the presentation, workup, and management of <i>SGCE</i> myoclonus-dystonia, a rare genetic condition, in a patient with atypical presenting symptoms and no family history of movement abnormalities.</p><p><strong>Methods: </strong>A woman with myoclonus and dystonia was identified based on clinical history and physical examination. Workup was conducted to determine the cause of her symptoms, including whole-exome sequencing. Myoclonus-dystonia is associated with more than 100 distinct mutations in MYC/DYT<i>-SGCE</i> that account for only half of the total myoclonus-dystonia patients. As such, this case required intensive genetic analyses rather than screening only for a small subset of well-characterized mutations.</p><p><strong>Results: </strong>Childhood onset myoclonus and worsening dystonia with age were identified in a young woman. She underwent screening for common causes of twitching movements, followed by whole-exome sequencing which identified a de novo novel variant in the <i>SGCE</i> gene, resulting in a diagnosis of <i>SGCE</i> myoclonus-dystonia.</p><p><strong>Discussion: </strong>Myoclonus-dystonia should be considered in patients with symptoms of head and upper extremity myoclonus early in life, especially with co-occurring dystonia, even in the absence of a family history of similar symptoms. Diagnosis of this condition should take place using sequencing, as new mutations continue to be discovered.</p>","PeriodicalId":48613,"journal":{"name":"Neurology-Genetics","volume":"10 2","pages":"e200128"},"PeriodicalIF":3.7000,"publicationDate":"2024-03-07","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC10932734/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Neurology-Genetics","FirstCategoryId":"3","ListUrlMain":"https://doi.org/10.1212/NXG.0000000000200128","RegionNum":3,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2024/4/1 0:00:00","PubModel":"eCollection","JCR":"Q2","JCRName":"CLINICAL NEUROLOGY","Score":null,"Total":0}
引用次数: 0
Abstract
Objectives: Characterize the presentation, workup, and management of SGCE myoclonus-dystonia, a rare genetic condition, in a patient with atypical presenting symptoms and no family history of movement abnormalities.
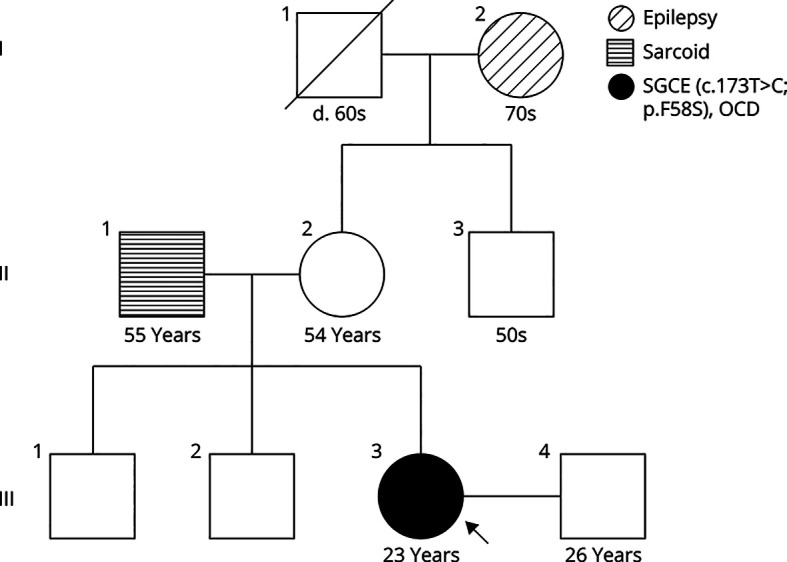
Methods: A woman with myoclonus and dystonia was identified based on clinical history and physical examination. Workup was conducted to determine the cause of her symptoms, including whole-exome sequencing. Myoclonus-dystonia is associated with more than 100 distinct mutations in MYC/DYT-SGCE that account for only half of the total myoclonus-dystonia patients. As such, this case required intensive genetic analyses rather than screening only for a small subset of well-characterized mutations.
Results: Childhood onset myoclonus and worsening dystonia with age were identified in a young woman. She underwent screening for common causes of twitching movements, followed by whole-exome sequencing which identified a de novo novel variant in the SGCE gene, resulting in a diagnosis of SGCE myoclonus-dystonia.
Discussion: Myoclonus-dystonia should be considered in patients with symptoms of head and upper extremity myoclonus early in life, especially with co-occurring dystonia, even in the absence of a family history of similar symptoms. Diagnosis of this condition should take place using sequencing, as new mutations continue to be discovered.
期刊介绍:
Neurology: Genetics is an online open access journal publishing peer-reviewed reports in the field of neurogenetics. Original articles in all areas of neurogenetics will be published including rare and common genetic variation, genotype-phenotype correlations, outlier phenotypes as a result of mutations in known disease-genes, and genetic variations with a putative link to diseases. This will include studies reporting on genetic disease risk and pharmacogenomics. In addition, Neurology: Genetics will publish results of gene-based clinical trials (viral, ASO, etc.). Genetically engineered model systems are not a primary focus of Neurology: Genetics, but studies using model systems for treatment trials are welcome, including well-powered studies reporting negative results.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
