Roberta De Mori, Silvia Tardivo, Lidia Pollara, Silvia Clara Giliani, Eltahir Ali, Lucio Giordano, Vincenzo Leuzzi, Rita Fischetto, Blanca Gener, Santo Diprima, Marco J. Morelli, Maria Cristina Monti, Virginie Sottile, Enza Maria Valente
{"title":"Joubert syndrome-derived induced pluripotent stem cells show altered neuronal differentiation in vitro","authors":"Roberta De Mori, Silvia Tardivo, Lidia Pollara, Silvia Clara Giliani, Eltahir Ali, Lucio Giordano, Vincenzo Leuzzi, Rita Fischetto, Blanca Gener, Santo Diprima, Marco J. Morelli, Maria Cristina Monti, Virginie Sottile, Enza Maria Valente","doi":"10.1007/s00441-024-03876-9","DOIUrl":null,"url":null,"abstract":"<p>Joubert syndrome (JS) is a recessively inherited congenital ataxia characterized by hypotonia, psychomotor delay, abnormal ocular movements, intellectual disability, and a peculiar cerebellar and brainstem malformation, the “molar tooth sign.” Over 40 causative genes have been reported, all encoding for proteins implicated in the structure or functioning of the primary cilium, a subcellular organelle widely present in embryonic and adult tissues. In this paper, we developed an <i>in vitro</i> neuronal differentiation model using patient-derived induced pluripotent stem cells (iPSCs), to evaluate possible neurodevelopmental defects in JS. To this end, iPSCs from four JS patients harboring mutations in distinct JS genes (<i>AHI1, CPLANE1</i>, <i>TMEM67</i>, and <i>CC2D2A</i>) were differentiated alongside healthy control cells to obtain mid-hindbrain precursors and cerebellar granule cells. Differentiation was monitored over 31 days through the detection of lineage-specific marker expression by qRT-PCR, immunofluorescence, and transcriptomics analysis. All JS patient-derived iPSCs, regardless of the mutant gene, showed a similar impairment to differentiate into mid-hindbrain and cerebellar granule cells when compared to healthy controls. In addition, analysis of primary cilium count and morphology showed notable ciliary defects in all differentiating JS patient-derived iPSCs compared to controls. These results confirm that patient-derived iPSCs are an accessible and relevant <i>in vitro</i> model to analyze cellular phenotypes connected to the presence of JS gene mutations in a neuronal context.</p>","PeriodicalId":9712,"journal":{"name":"Cell and Tissue Research","volume":"30 1","pages":""},"PeriodicalIF":2.9000,"publicationDate":"2024-03-19","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Cell and Tissue Research","FirstCategoryId":"99","ListUrlMain":"https://doi.org/10.1007/s00441-024-03876-9","RegionNum":3,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q3","JCRName":"CELL BIOLOGY","Score":null,"Total":0}
引用次数: 0
Abstract
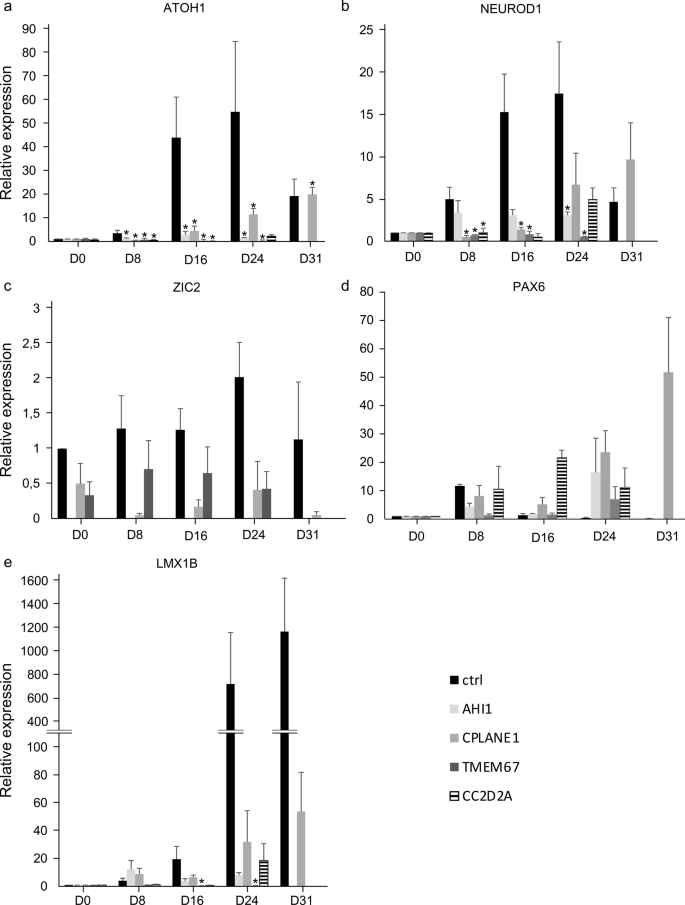
Joubert syndrome (JS) is a recessively inherited congenital ataxia characterized by hypotonia, psychomotor delay, abnormal ocular movements, intellectual disability, and a peculiar cerebellar and brainstem malformation, the “molar tooth sign.” Over 40 causative genes have been reported, all encoding for proteins implicated in the structure or functioning of the primary cilium, a subcellular organelle widely present in embryonic and adult tissues. In this paper, we developed an in vitro neuronal differentiation model using patient-derived induced pluripotent stem cells (iPSCs), to evaluate possible neurodevelopmental defects in JS. To this end, iPSCs from four JS patients harboring mutations in distinct JS genes (AHI1, CPLANE1, TMEM67, and CC2D2A) were differentiated alongside healthy control cells to obtain mid-hindbrain precursors and cerebellar granule cells. Differentiation was monitored over 31 days through the detection of lineage-specific marker expression by qRT-PCR, immunofluorescence, and transcriptomics analysis. All JS patient-derived iPSCs, regardless of the mutant gene, showed a similar impairment to differentiate into mid-hindbrain and cerebellar granule cells when compared to healthy controls. In addition, analysis of primary cilium count and morphology showed notable ciliary defects in all differentiating JS patient-derived iPSCs compared to controls. These results confirm that patient-derived iPSCs are an accessible and relevant in vitro model to analyze cellular phenotypes connected to the presence of JS gene mutations in a neuronal context.
期刊介绍:
The journal publishes regular articles and reviews in the areas of molecular, cell, and supracellular biology. In particular, the journal intends to provide a forum for publishing data that analyze the supracellular, integrative actions of gene products and their impact on the formation of tissue structure and function. Submission of papers with an emphasis on structure-function relationships as revealed by recombinant molecular technologies is especially encouraged. Areas of research with a long-standing tradition of publishing in Cell & Tissue Research include:
- neurobiology
- neuroendocrinology
- endocrinology
- reproductive biology
- skeletal and immune systems
- development
- stem cells
- muscle biology.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
