Romdhane Rekaya, Shannon Smith, El Hamidi Hay, Nourhene Farhat, Samuel E Aggrey
{"title":"Analysis of binary responses with outcome-specific misclassification probability in genome-wide association studies.","authors":"Romdhane Rekaya, Shannon Smith, El Hamidi Hay, Nourhene Farhat, Samuel E Aggrey","doi":"10.2147/TACG.S122250","DOIUrl":null,"url":null,"abstract":"<p><p>Errors in the binary status of some response traits are frequent in human, animal, and plant applications. These error rates tend to differ between cases and controls because diagnostic and screening tests have different sensitivity and specificity. This increases the inaccuracies of classifying individuals into correct groups, giving rise to both false-positive and false-negative cases. The analysis of these noisy binary responses due to misclassification will undoubtedly reduce the statistical power of genome-wide association studies (GWAS). A threshold model that accommodates varying diagnostic errors between cases and controls was investigated. A simulation study was carried out where several binary data sets (case-control) were generated with varying effects for the most influential single nucleotide polymorphisms (SNPs) and different diagnostic error rate for cases and controls. Each simulated data set consisted of 2000 individuals. Ignoring misclassification resulted in biased estimates of true influential SNP effects and inflated estimates for true noninfluential markers. A substantial reduction in bias and increase in accuracy ranging from 12% to 32% was observed when the misclassification procedure was invoked. In fact, the majority of influential SNPs that were not identified using the noisy data were captured using the proposed method. Additionally, truly misclassified binary records were identified with high probability using the proposed method. The superiority of the proposed method was maintained across different simulation parameters (misclassification rates and odds ratios) attesting to its robustness.</p>","PeriodicalId":39131,"journal":{"name":"Application of Clinical Genetics","volume":"9 ","pages":"169-177"},"PeriodicalIF":2.6000,"publicationDate":"2016-11-30","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5138056/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Application of Clinical Genetics","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.2147/TACG.S122250","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2016/1/1 0:00:00","PubModel":"eCollection","JCR":"Q2","JCRName":"GENETICS & HEREDITY","Score":null,"Total":0}
引用次数: 0
Abstract
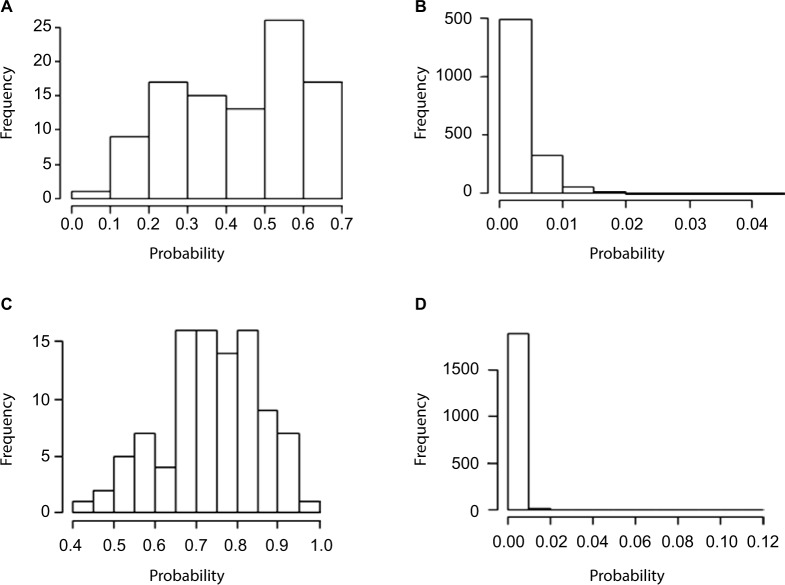
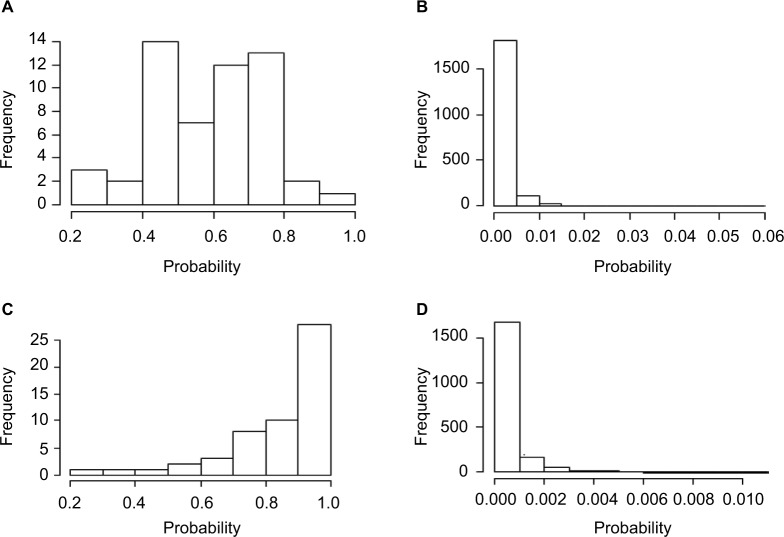
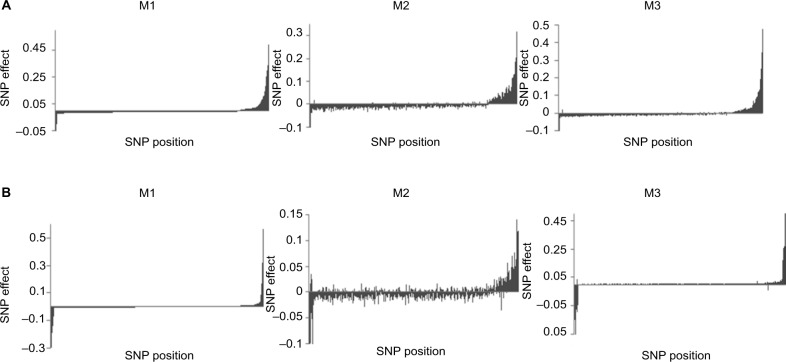
Errors in the binary status of some response traits are frequent in human, animal, and plant applications. These error rates tend to differ between cases and controls because diagnostic and screening tests have different sensitivity and specificity. This increases the inaccuracies of classifying individuals into correct groups, giving rise to both false-positive and false-negative cases. The analysis of these noisy binary responses due to misclassification will undoubtedly reduce the statistical power of genome-wide association studies (GWAS). A threshold model that accommodates varying diagnostic errors between cases and controls was investigated. A simulation study was carried out where several binary data sets (case-control) were generated with varying effects for the most influential single nucleotide polymorphisms (SNPs) and different diagnostic error rate for cases and controls. Each simulated data set consisted of 2000 individuals. Ignoring misclassification resulted in biased estimates of true influential SNP effects and inflated estimates for true noninfluential markers. A substantial reduction in bias and increase in accuracy ranging from 12% to 32% was observed when the misclassification procedure was invoked. In fact, the majority of influential SNPs that were not identified using the noisy data were captured using the proposed method. Additionally, truly misclassified binary records were identified with high probability using the proposed method. The superiority of the proposed method was maintained across different simulation parameters (misclassification rates and odds ratios) attesting to its robustness.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
