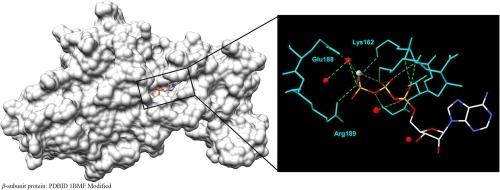
{"title":"Structural analysis of ATP bound to the F1-ATPase β-subunit monomer by solid-state NMR- insight into the hydrolysis mechanism in F1","authors":"Yasuto Todokoro , Yoshiyuki Miyasaka , Hiromasa Yagi , Masatsune Kainosho , Toshimichi Fujiwara , Hideo Akutsu","doi":"10.1016/j.bpc.2024.107232","DOIUrl":null,"url":null,"abstract":"<div><p>ATP-hydrolysis-associated conformational change of the <em>β</em>-subunit during the rotation of F<sub>1</sub>-ATPase (F<sub>1</sub>) has been discussed using cryo-electron microscopy (cryo-EM). Since it is worthwhile to further investigate the conformation of ATP at the catalytic subunit through an alternative approach, the structure of ATP bound to the F<sub>1</sub><em>β</em>-subunit monomer (<em>β</em>) was analyzed by solid-state NMR. The adenosine conformation of ATP-<em>β</em> was similar to that of ATP analog in F<sub>1</sub> crystal structures. <sup>31</sup>P chemical shift analysis showed that the P<sup>α</sup> and P<sup>β</sup> conformations of ATP-<em>β</em> are gauche-trans and trans-trans, respectively. The triphosphate chain is more extended in ATP-<em>β</em> than in ATP analog in F<sub>1</sub> crystals. This appears to be in the state just before ATP hydrolysis. Furthermore, the ATP-<em>β</em> conformation is known to be more closed than the closed form in F<sub>1</sub> crystal structures. In view of the cryo-EM results, ATP-<em>β</em> would be a model of the most closed <em>β</em>-subunit with ATP ready for hydrolysis in the hydrolysis stroke of the F<sub>1</sub> rotation.</p></div>","PeriodicalId":8979,"journal":{"name":"Biophysical chemistry","volume":"309 ","pages":"Article 107232"},"PeriodicalIF":2.2000,"publicationDate":"2024-04-03","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Biophysical chemistry","FirstCategoryId":"99","ListUrlMain":"https://www.sciencedirect.com/science/article/pii/S0301462224000619","RegionNum":3,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"BIOCHEMISTRY & MOLECULAR BIOLOGY","Score":null,"Total":0}
引用次数: 0
Abstract
ATP-hydrolysis-associated conformational change of the β-subunit during the rotation of F1-ATPase (F1) has been discussed using cryo-electron microscopy (cryo-EM). Since it is worthwhile to further investigate the conformation of ATP at the catalytic subunit through an alternative approach, the structure of ATP bound to the F1β-subunit monomer (β) was analyzed by solid-state NMR. The adenosine conformation of ATP-β was similar to that of ATP analog in F1 crystal structures. 31P chemical shift analysis showed that the Pα and Pβ conformations of ATP-β are gauche-trans and trans-trans, respectively. The triphosphate chain is more extended in ATP-β than in ATP analog in F1 crystals. This appears to be in the state just before ATP hydrolysis. Furthermore, the ATP-β conformation is known to be more closed than the closed form in F1 crystal structures. In view of the cryo-EM results, ATP-β would be a model of the most closed β-subunit with ATP ready for hydrolysis in the hydrolysis stroke of the F1 rotation.
通过固态核磁共振分析与 F1-ATP 酶 β 亚基单体结合的 ATP 结构--深入了解 F1 的水解机制
利用低温电子显微镜(cryo-EM)讨论了F1-ATP酶(F1)旋转过程中β亚基与ATP水解相关的构象变化。由于值得通过另一种方法进一步研究 ATP 在催化亚基上的构象,因此采用固态核磁共振分析了与 F1β 亚基单体(β)结合的 ATP 结构。ATP-β 的腺苷构象与 F1 晶体结构中的 ATP 类似物相似。31P 化学位移分析表明,ATP-β 的 Pα 和 Pβ 构象分别为高-反式和反-反式。与 F1 晶体中的 ATP 类似物相比,ATP-β 中的三磷酸链延伸得更长。这似乎是 ATP 水解前的状态。此外,已知 ATP-β 的构象比 F1 晶体结构中的封闭形式更为封闭。鉴于低温电子显微镜的结果,ATP-β 将是最封闭的 β 亚基模型,在 F1 旋转的水解行程中,ATP 已准备好进行水解。
期刊介绍:
Biophysical Chemistry publishes original work and reviews in the areas of chemistry and physics directly impacting biological phenomena. Quantitative analysis of the properties of biological macromolecules, biologically active molecules, macromolecular assemblies and cell components in terms of kinetics, thermodynamics, spatio-temporal organization, NMR and X-ray structural biology, as well as single-molecule detection represent a major focus of the journal. Theoretical and computational treatments of biomacromolecular systems, macromolecular interactions, regulatory control and systems biology are also of interest to the journal.


 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
