Genome-wide analysis identifies MYH11 compound heterozygous variants leading to visceral myopathy corresponding to late-onset form of megacystis-microcolon-intestinal hypoperistalsis syndrome
Clarisse Billon, Giorgina Barbara Piccoli, Jean-Madeleine de Sainte Agathe, Radka Stoeva, Nicolas Derive, Laurence Heidet, Dominique Berrebi, Patrick Bruneval, Xavier Jeunemaitre, Marguerite Hureaux
{"title":"Genome-wide analysis identifies MYH11 compound heterozygous variants leading to visceral myopathy corresponding to late-onset form of megacystis-microcolon-intestinal hypoperistalsis syndrome","authors":"Clarisse Billon, Giorgina Barbara Piccoli, Jean-Madeleine de Sainte Agathe, Radka Stoeva, Nicolas Derive, Laurence Heidet, Dominique Berrebi, Patrick Bruneval, Xavier Jeunemaitre, Marguerite Hureaux","doi":"10.1007/s00438-024-02136-3","DOIUrl":null,"url":null,"abstract":"<p>Megacystis-microcolon-hypoperistalsis-syndrome (MMIHS) is a rare and early-onset congenital disease characterized by massive abdominal distension due to a large non-obstructive bladder, a microcolon and decreased or absent intestinal peristalsis. While in most cases inheritance is autosomal dominant and associated with heterozygous variant in <i>ACTG2</i> gene, an autosomal recessive transmission has also been described including pathogenic bialellic loss-of-function variants in <i>MYH11</i>. We report here a novel family with visceral myopathy related to <i>MYH11</i> gene, confirmed by whole genome sequencing (WGS). WGS was performed in two siblings with unusual presentation of MMIHS and their two healthy parents. The 38 years-old brother had severe bladder dysfunction and intestinal obstruction, whereas the 30 years-old sister suffered from end-stage kidney disease with neurogenic bladder and recurrent sigmoid volvulus. WGS was completed by retrospective digestive pathological analyses. Compound heterozygous variants of <i>MYH11</i> gene were identified, associating a deletion of 1.2 Mb encompassing <i>MYH11</i> inherited from the father and an in-frame variant c.2578_2580del, p.Glu860del inherited from the mother. Pathology analyses of the colon and the rectum revealed structural changes which significance of which is discussed. Cardiac and vascular assessment of the mother was normal. This is the second report of a visceral myopathy corresponding to late-onset form of MMIHS related to compound heterozygosity in <i>MYH11;</i> with complete gene deletion and a hypomorphic allele <i>in trans</i>. The hypomorphic allele harbored by the mother raised the question of the risk of aortic disease in adults. This case shows the interest of WGS in deciphering complex phenotypes, allowing adapted diagnosis and genetic counselling.</p>","PeriodicalId":18816,"journal":{"name":"Molecular Genetics and Genomics","volume":"72 1","pages":""},"PeriodicalIF":2.1000,"publicationDate":"2024-04-16","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Molecular Genetics and Genomics","FirstCategoryId":"99","ListUrlMain":"https://doi.org/10.1007/s00438-024-02136-3","RegionNum":3,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q3","JCRName":"BIOCHEMISTRY & MOLECULAR BIOLOGY","Score":null,"Total":0}
引用次数: 0
Abstract
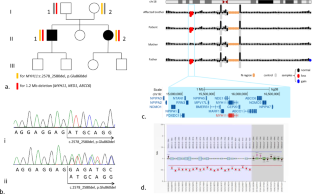
Megacystis-microcolon-hypoperistalsis-syndrome (MMIHS) is a rare and early-onset congenital disease characterized by massive abdominal distension due to a large non-obstructive bladder, a microcolon and decreased or absent intestinal peristalsis. While in most cases inheritance is autosomal dominant and associated with heterozygous variant in ACTG2 gene, an autosomal recessive transmission has also been described including pathogenic bialellic loss-of-function variants in MYH11. We report here a novel family with visceral myopathy related to MYH11 gene, confirmed by whole genome sequencing (WGS). WGS was performed in two siblings with unusual presentation of MMIHS and their two healthy parents. The 38 years-old brother had severe bladder dysfunction and intestinal obstruction, whereas the 30 years-old sister suffered from end-stage kidney disease with neurogenic bladder and recurrent sigmoid volvulus. WGS was completed by retrospective digestive pathological analyses. Compound heterozygous variants of MYH11 gene were identified, associating a deletion of 1.2 Mb encompassing MYH11 inherited from the father and an in-frame variant c.2578_2580del, p.Glu860del inherited from the mother. Pathology analyses of the colon and the rectum revealed structural changes which significance of which is discussed. Cardiac and vascular assessment of the mother was normal. This is the second report of a visceral myopathy corresponding to late-onset form of MMIHS related to compound heterozygosity in MYH11; with complete gene deletion and a hypomorphic allele in trans. The hypomorphic allele harbored by the mother raised the question of the risk of aortic disease in adults. This case shows the interest of WGS in deciphering complex phenotypes, allowing adapted diagnosis and genetic counselling.
期刊介绍:
Molecular Genetics and Genomics (MGG) publishes peer-reviewed articles covering all areas of genetics and genomics. Any approach to the study of genes and genomes is considered, be it experimental, theoretical or synthetic. MGG publishes research on all organisms that is of broad interest to those working in the fields of genetics, genomics, biology, medicine and biotechnology.
The journal investigates a broad range of topics, including these from recent issues: mechanisms for extending longevity in a variety of organisms; screening of yeast metal homeostasis genes involved in mitochondrial functions; molecular mapping of cultivar-specific avirulence genes in the rice blast fungus and more.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
