{"title":"In silico anticancer activity of isoxazolidine and isoxazolines derivatives: DFT study, ADMET prediction, and molecular docking","authors":"Moulay Driss Mellaoui , Khadija Zaki , Khalid Abbiche , Abdallah Imjjad , Rachid Boutiddar , Abdelouahid Sbai , Aaziz Jmiai , Souad El Issami , Al Mokhtar Lamsabhi , Hanane Zejli","doi":"10.1016/j.molstruc.2024.138330","DOIUrl":null,"url":null,"abstract":"<div><p>This study presents a comprehensive analysis of six isoxazolidine and isoxazoline derivatives, employing a multifaceted approach that integrates Density Functional Theory (DFT), AdmetSAR analysis, and molecular docking simulations to explore their electronic, pharmacokinetic, and anticancer properties. Utilizing DFT analysis with the B3LYP-D3BJ functional and the 6-311++G(d,p) basis set, molecular geometries were optimized, and vibrational frequencies in the IR spectrum were evaluated, offering insights into the molecular structure and stability of the pharmaceutical compounds. Electrostatic potential maps were analyzed to predict functional group reactivity and protein-substrate interactions. Frontier Molecular Orbital (FMO) analysis and Density of States (DOS) plots revealed varying stability levels among the compounds, with 1b, 2b, and 3b exhibiting slightly higher stability. Chemical potential and hardness analyses highlighted stronger binding affinity for compounds 1b and 2b, suggesting stronger potential interactions. AdmetSAR analysis predicted favorable human intestinal absorption (HIA) rates for all compounds, with compound 3b showing superior oral effectiveness. Molecular docking and dynamics simulations were conducted targeting the receptor (PDB: 1JU6). Molecular docking simulations confirmed the high affinity of these compounds towards the target protein 1JU6, particularly compound 3b, which exhibited the most favorable binding energy of -8.50 kcal/mol. Molecular dynamics simulations demonstrated the superior stability of ligand 3b over 1b and 5-FU over 100 ns, suggesting its potential for further study. The 3b-protein complex exhibited stability through hydrophobic and hydrogen bond interactions, with 3b demonstrating reduced solvent exposure compared to 1b and 5-FU. This study underscores the promising role of compound 3b in anticancer treatments, providing a solid foundation for future drug development and optimization efforts.</p></div>","PeriodicalId":16414,"journal":{"name":"Journal of Molecular Structure","volume":"1308 ","pages":"Article 138330"},"PeriodicalIF":4.7000,"publicationDate":"2024-07-15","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Molecular Structure","FirstCategoryId":"92","ListUrlMain":"https://www.sciencedirect.com/science/article/pii/S0022286024008500","RegionNum":2,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2024/4/12 0:00:00","PubModel":"Epub","JCR":"Q2","JCRName":"CHEMISTRY, PHYSICAL","Score":null,"Total":0}
引用次数: 0
Abstract
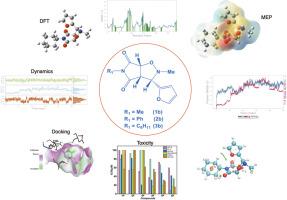
This study presents a comprehensive analysis of six isoxazolidine and isoxazoline derivatives, employing a multifaceted approach that integrates Density Functional Theory (DFT), AdmetSAR analysis, and molecular docking simulations to explore their electronic, pharmacokinetic, and anticancer properties. Utilizing DFT analysis with the B3LYP-D3BJ functional and the 6-311++G(d,p) basis set, molecular geometries were optimized, and vibrational frequencies in the IR spectrum were evaluated, offering insights into the molecular structure and stability of the pharmaceutical compounds. Electrostatic potential maps were analyzed to predict functional group reactivity and protein-substrate interactions. Frontier Molecular Orbital (FMO) analysis and Density of States (DOS) plots revealed varying stability levels among the compounds, with 1b, 2b, and 3b exhibiting slightly higher stability. Chemical potential and hardness analyses highlighted stronger binding affinity for compounds 1b and 2b, suggesting stronger potential interactions. AdmetSAR analysis predicted favorable human intestinal absorption (HIA) rates for all compounds, with compound 3b showing superior oral effectiveness. Molecular docking and dynamics simulations were conducted targeting the receptor (PDB: 1JU6). Molecular docking simulations confirmed the high affinity of these compounds towards the target protein 1JU6, particularly compound 3b, which exhibited the most favorable binding energy of -8.50 kcal/mol. Molecular dynamics simulations demonstrated the superior stability of ligand 3b over 1b and 5-FU over 100 ns, suggesting its potential for further study. The 3b-protein complex exhibited stability through hydrophobic and hydrogen bond interactions, with 3b demonstrating reduced solvent exposure compared to 1b and 5-FU. This study underscores the promising role of compound 3b in anticancer treatments, providing a solid foundation for future drug development and optimization efforts.
期刊介绍:
The Journal of Molecular Structure is dedicated to the publication of full-length articles and review papers, providing important new structural information on all types of chemical species including:
• Stable and unstable molecules in all types of environments (vapour, molecular beam, liquid, solution, liquid crystal, solid state, matrix-isolated, surface-absorbed etc.)
• Chemical intermediates
• Molecules in excited states
• Biological molecules
• Polymers.
The methods used may include any combination of spectroscopic and non-spectroscopic techniques, for example:
• Infrared spectroscopy (mid, far, near)
• Raman spectroscopy and non-linear Raman methods (CARS, etc.)
• Electronic absorption spectroscopy
• Optical rotatory dispersion and circular dichroism
• Fluorescence and phosphorescence techniques
• Electron spectroscopies (PES, XPS), EXAFS, etc.
• Microwave spectroscopy
• Electron diffraction
• NMR and ESR spectroscopies
• Mössbauer spectroscopy
• X-ray crystallography
• Charge Density Analyses
• Computational Studies (supplementing experimental methods)
We encourage publications combining theoretical and experimental approaches. The structural insights gained by the studies should be correlated with the properties, activity and/ or reactivity of the molecule under investigation and the relevance of this molecule and its implications should be discussed.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
