Study of ab initio calculations of structural, electronic and optical properties of ternary semiconductor \({\mathbf{G}\mathbf{a}}_{1-\mathbf{x}}{\mathbf{I}\mathbf{n}}_{\mathbf{x}}\mathbf{S}\mathbf{b}\) alloys
{"title":"Study of ab initio calculations of structural, electronic and optical properties of ternary semiconductor \\({\\mathbf{G}\\mathbf{a}}_{1-\\mathbf{x}}{\\mathbf{I}\\mathbf{n}}_{\\mathbf{x}}\\mathbf{S}\\mathbf{b}\\) alloys","authors":"Maryam Noorafshan, Sina Heydari","doi":"10.1007/s12034-024-03177-5","DOIUrl":null,"url":null,"abstract":"<div><p>First principles calculations of the structural, electronic and optical properties of GaSb, InSb and their ternary <span>\\({{\\text{Ga}}}_{1-x}{{\\text{In}}}_{x}{\\text{Sb}}\\)</span> alloys (<i>x</i> = 0.25, 0.50 and 0.75) have been performed using the full-potential linear muffin-tin orbital (FP-LAPW) method within density functional theory (DFT). The generalized gradient approximation and modified Becke and Janson functional with local density approximation (mBJ–LDA) are utilized for the treatment of exchange and correlation potentials. The results show the calculated lattice constants increase linearity with increasing the In concentration. The electronic band structure indicates that these alloys are direct band gap semiconductors for all the values of <i>x</i> and the band gap decreases as <i>x</i> increases from <i>x</i> = 0 to <i>x</i> = 1. The results also show that the there is a non-linear dependence of the band gap on composition <i>x</i> in <span>\\({{\\text{Ga}}}_{1-x}{{\\text{In}}}_{x}{\\text{Sb}}\\)</span> alloys. Regarding optical properties, the real and imaginary parts of the dielectric function have been calculated. The results show that the static dielectric constant increases with increase in concentration of In, consistent with reduction in energy band gap. The onset point and major peaks in the imaginary parts of the dielectric function spectra are identified for the <span>\\({{\\text{Ga}}}_{1-x}{{\\text{In}}}_{x}{\\text{Sb}}\\)</span> alloys and shown that they are related to the corresponding band gap values. The results also show that for energy values higher than <span>\\(\\cong 4\\)</span> eV, the electromagnetic wave transition through <span>\\({{\\text{Ga}}}_{1-x}{{\\text{In}}}_{x}{\\text{Sb}}\\)</span> alloys is nearly zero.</p></div>","PeriodicalId":502,"journal":{"name":"Bulletin of Materials Science","volume":"47 2","pages":""},"PeriodicalIF":2.1000,"publicationDate":"2024-04-23","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Bulletin of Materials Science","FirstCategoryId":"88","ListUrlMain":"https://link.springer.com/article/10.1007/s12034-024-03177-5","RegionNum":4,"RegionCategory":"材料科学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q3","JCRName":"MATERIALS SCIENCE, MULTIDISCIPLINARY","Score":null,"Total":0}
引用次数: 0
Abstract
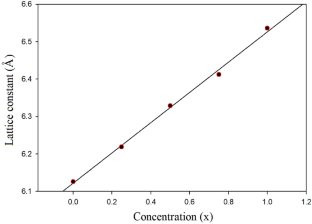
First principles calculations of the structural, electronic and optical properties of GaSb, InSb and their ternary \({{\text{Ga}}}_{1-x}{{\text{In}}}_{x}{\text{Sb}}\) alloys (x = 0.25, 0.50 and 0.75) have been performed using the full-potential linear muffin-tin orbital (FP-LAPW) method within density functional theory (DFT). The generalized gradient approximation and modified Becke and Janson functional with local density approximation (mBJ–LDA) are utilized for the treatment of exchange and correlation potentials. The results show the calculated lattice constants increase linearity with increasing the In concentration. The electronic band structure indicates that these alloys are direct band gap semiconductors for all the values of x and the band gap decreases as x increases from x = 0 to x = 1. The results also show that the there is a non-linear dependence of the band gap on composition x in \({{\text{Ga}}}_{1-x}{{\text{In}}}_{x}{\text{Sb}}\) alloys. Regarding optical properties, the real and imaginary parts of the dielectric function have been calculated. The results show that the static dielectric constant increases with increase in concentration of In, consistent with reduction in energy band gap. The onset point and major peaks in the imaginary parts of the dielectric function spectra are identified for the \({{\text{Ga}}}_{1-x}{{\text{In}}}_{x}{\text{Sb}}\) alloys and shown that they are related to the corresponding band gap values. The results also show that for energy values higher than \(\cong 4\) eV, the electromagnetic wave transition through \({{\text{Ga}}}_{1-x}{{\text{In}}}_{x}{\text{Sb}}\) alloys is nearly zero.
三元半导体 $${{mathbf{G}\mathbf{a}}_{{1-\mathbf{x}}{mathbf{I}\mathbf{n}}_{{mathbf{x}}\mathbf{S}$ 合金的结构、电子和光学特性的 ab initio 计算研究
使用密度泛函理论(DFT)中的全电位线性松饼-锡轨道(FP-LAPW)方法,对 GaSb、InSb 及其三元合金(x = 0.25、0.50 和 0.75)的结构、电子和光学性质进行了第一性原理计算。在处理交换势和相关势时,使用了广义梯度近似和局部密度近似的修正贝克和扬森函数(mBJ-LDA)。结果表明,随着铟浓度的增加,计算得到的晶格常数呈线性增长。电子带结构表明,这些合金在所有 x 值下都是直接带隙半导体,带隙随着 x 值从 x = 0 到 x = 1 的增加而减小。结果还表明,在 \({{text{Ga}}}_{1-x}{{text{In}}}_{x}{text{Sb}}/)合金中,带隙与成分 x 存在非线性依赖关系。在光学特性方面,计算了介电函数的实部和虚部。结果表明,静态介电常数随着铟浓度的增加而增大,这与能带隙的减小是一致的。确定了 \({{text{Ga}}}_{1-x}{{text{In}}}_{x}{text{Sb}}\)合金介电函数谱虚部的起始点和主要峰值,并表明它们与相应的带隙值有关。结果还表明,当能量值高于 \(\cong 4\) eV 时,通过 \({{text{Ga}}_{1-x}{{text{In}}}_{x}{text{Sb}}\) 合金的电磁波跃迁几乎为零。
期刊介绍:
The Bulletin of Materials Science is a bi-monthly journal being published by the Indian Academy of Sciences in collaboration with the Materials Research Society of India and the Indian National Science Academy. The journal publishes original research articles, review articles and rapid communications in all areas of materials science. The journal also publishes from time to time important Conference Symposia/ Proceedings which are of interest to materials scientists. It has an International Advisory Editorial Board and an Editorial Committee. The Bulletin accords high importance to the quality of articles published and to keep at a minimum the processing time of papers submitted for publication.




 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
