{"title":"Dietary long-chain fatty acid metabolism boosts antitumor immune response","authors":"Jiaming Wang, Xuetao Cao","doi":"10.1002/cac2.12543","DOIUrl":null,"url":null,"abstract":"<p>Overcoming resistance to immune checkpoint blockade (ICB) therapy will pave the way for effective ICB cancer immunotherapy since a large proportion of cancer patients are not responsive to ICB immunotherapy [<span>1</span>]. The molecular mechanisms of ICB resistance are diverse, including tumor-intrinsic resistant factors (such as genetic and epigenetic disorders), immunosuppressive/disabled factors (such as T cell exhaustion), and environmental restrictive factors (such as neuroendocrine stress and metabolic reprogramming). Deciphering the mechanisms for ICB resistance will provide immense potential for designing new immunotherapeutic strategies in refractory cancers. The tumor microenvironment (TME) includes diverse types of cells, such as immune cells, cancer-associated fibroblasts, and endothelial cells, as well as intercellular cytokines/chemokines, growth factors, and metabolites, which have been recognized as crucial determinants in ICB responsiveness. The recent advent of high-throughput metabolomics and lipidomics analysis reveals that metabolic reprogramming in TME is closely associated with cancer cell invasion, regulated cell death, immune escape, and chemoresistance [<span>2</span>]. On the other hand, immunometabolism also modulates tumor-associated immune cell function and immunotherapy efficacy. For instance, higher expression of major facilitator superfamily domain containing 2A (MFSD2A) in gastric cancer cells inhibits transforming growth factor beta 1 (TGF-β1) production by suppressing cyclooxygenase 2 (COX2)-prostaglandin synthesis, thus promoting antitumor immunity via reprogramming TME [<span>3</span>].</p><p>As a major component of lipids in TME, long-chain fatty acids (LCFAs) are important energy supply and cellular membrane components for cancer cells. Indeed, various types of LCFAs have been found in TME, showing different and sometimes opposite influences on tumor progression and antitumor immunity [<span>4</span>]. For example, palmitic acid promotes metastasis in oral carcinomas and melanoma mouse models through stimulating intratumoral Schwann cells and innervation [<span>5</span>], whereas linoleic acid potentiates CD8<sup>+</sup> T cell antitumor functions via enhancing endoplasmic reticulum-mitochondria contact formation and energetics fitness [<span>6</span>]. Therefore, better understanding of the molecular mechanism for each individual LCFA in tumor immunity is meaningful since it may improve cancer immunotherapy through targeting metabolic reprogramming of TME. In a recent study published in <i>Cell Metabolism</i>, Lai <i>et al.</i> [<span>7</span>] found dietary elaidic acid (EA) supplementation elevates tumoral major histocompatibility complex-1 (MHC-I) expression via acyl-coenzyme A synthetase long chain family member 5 (ACSL5), thus suppressing tumor growth and enhancing anti-programmed cell death protein 1 (anti-PD-1) efficacy (Figure 1).</p><p>Acyl-coenzyme A synthetase long-chain family members (ACSLs) catalyze the conversion of LCFAs into LCFA-coenzyme A (LCFA-CoA) for further lipid catabolism or anabolism. It is reported that interferon gamma (IFNγ) released by cytotoxic T lymphocytes (CTLs) and arachidonic acid in TME coordinately induce tumor cell ferroptosis via acyl-coenzyme A synthetase long chain family member 4 (ACSL4) [<span>8</span>]. Therefore, manipulating ACSLs activity might be an ideal target for metabolic reprogramming in cancer therapy. In this study, the authors found ectopic expression of acyl-coenzyme A synthetase long chain family member 1 (ACSL1), ACSL5, or acyl-coenzyme A synthetase long chain family member (ACSL6) in B16-F10 melanoma cells reduced tumor growth in a subcutaneous mouse model [<span>7</span>]. Given that higher levels of ACSL5 and ACSL6 mRNA were associated with improved overall survival in melanoma patients according to The Cancer Genome Atlas (TCGA) datasets and broad expression of ACSL5 in multiple tissues, the authors focused on ACSL5 for further study. ACSL5 expression also suppressed subcutaneous growth of Lewis lung carcinoma (LLC), Hepa1-6 hepatic tumor, and tumor-model-derived cancer cell line (TDCL) in mouse models. Interestingly, ACSL5-mediated antitumor effect was dependent on acquired immunity since ACSL5 failed to regulate tumor growth in immunodeficient non-obese diabetic-<i>Prkdc</i><sup>scid</sup><i>IL2rg</i><sup>em1</sup>/Smoc (M-NSG) and <i>Rag2</i><sup>−/−</sup> mice or upon anti-CD8 antibody treatment in C57BL/6 mice. Furthermore, ACSL5-deficient TDCL tumors were not responsive to anti-PD-1 therapy in contrast to wide-type tumors, indicating that tumor cell ACSL5 determines the sensitivity to ICB immunotherapy.</p><p>To further delineate ACSL5 antitumor function, the authors examined the number and function of tumor-infiltrating T cells. The number of CD8<sup>+</sup> T cells or CD4 <sup>+</sup> T cells and their cytokines (tumor necrosis factor α [TNF-α] and IFNγ) production were not changed in ACSL5-deficient TDCL tumors. Besides, ACSL5 ectopic B16-F10 tumors had no effects on the number of T cells but slightly increased the TNF-α production by CD8<sup>+</sup> T cells. Although CD8<sup>+</sup> T cell number was increased in ACSL5-expressing LLC tumors, the number of CD4<sup>+</sup> T cells and cytokines production by T cells were not influenced. Therefore, the authors speculated that ACSL5 might regulate tumor sensitivity to T cell-mediated killing. Indeed, they found that ACSL5 deficiency rendered tumor cells more resistant to CD8<sup>+</sup> T cell-mediated cytotoxicity in the co-culture system in vitro. Insufficient tumor immunogenicity is a pivotal cause of ICB resistance since CTLs mediate tumor cell killing through T cell receptor (TCR) recognition of tumor antigen presentation by MHC-I molecules [<span>9</span>]. Accordingly, the authors found downregulation of MHC-I expression in ACSL5-deficient tumor cells. Moreover, knockout of β-2 microglobulin (β2m), a component of MHC-I, promoted tumor growth in control tumors, but not in ACSL5-deficient tumors in vivo, indicating ACSL5 mediates antitumor effect through MHC-I upregulation.</p><p>By transcriptomic analysis of wild-type and ACSL5-deficient cells, the authors noticed that NLR family CARD domain containing 5 (NLRC5), one of the key transcription activators of MHC-I, was reduced upon ACSL5 knockout. Silencing NLRC5 completely abolished the MHC-I upregulation by ACSL5 ectopic expression, suggesting the ACSL5-NLRC5-MHC-I axis in determining tumor cell immunogenicity for induction of antitumor immunity.</p><p>Given that ACSLs take part in LCFAs activation, whether any LCFAs supplementation phenocopies ACSL5's antitumor function? According to various LCFAs screening, the authors found EA, an oleic acid <i>trans</i> isomer and a common component of animal fats and vegetable oils, potently enhanced basal and IFNγ-stimulated MHC-I expression. ACSL5 or NLRC5 deficiency impaired the induction of MHC-I by EA, further validating the EA-ACSL5-NLRC5-MHC-I axis in metabolic regulation of antitumor immunity. Besides, intraperitoneal and intragastrical EA administration both suppressed B16-F10 tumor growth in vivo in an immune-dependent manner. Consistently, EA treatment in combination with anti-PD-1 therapy significantly reduced LLC tumor burden, which was resistant to ICB originally, underscoring the promising therapeutic potential of dietary EA supplementation in promoting responsiveness to ICB. Finally, the authors found that higher plasma EA level was correlated with patients with better response to ICB and improved progression-free survival, implying that plasma EA might be an ideal predictor for ICB effectiveness.</p><p>Although numerous studies suggest the harmful effect of <i>trans</i> fatty acids (TFAs) especially in cardiovascular diseases, this study unveiled an unexpected role of TFAs in boosting tumor immunogenicity and enhancing antitumor immune response. However, there are still many questions that need to be addressed. How the metabolic enzyme ACSL5 regulates nuclear transcription factor NLRC5 expression, and how NLRC5 acts as a fatty acid metabolism alteration sensor are unclear. In contrast to this finding, EA has been reported to promote the growth and metastasis of mouse colorectal cancer cells by activating Wnt and extracellular signal-regulated kinase (ERK) signaling [<span>10</span>]. Whether the opposite functions of EA in different tumor models are due to TME heterogeneity needs further investigation. Although the authors focused on the antitumor effect of EA in tumor cells, whether EA also directly affects CD8<sup>+</sup> T cells function is unclear. It is also interesting to examine the lipid metabolism rewiring, fatty acid-mediated signaling transduction and antitumor function modulation in immune cells during EA intake. The authors demonstrated that ACSL5 converted EA into EA-CoA using isotype labeling, however, whether EA-CoA stimulated MHC-I expression directly through regulating target protein acylation like palmitoylation remains elusive. After conversion to EA-CoA, the activated fatty acids were essential for the downstream lipid metabolic pathways, such as fatty acid β-oxidation (FAO), lipid biosynthesis, and protein acylation (Figure 1). Our unpublished data suggest that ACSL5 can regulate phospholipid synthesis and cellular membrane properties, which might also impact membrane translocation of MHC-1 in tumor cells and other receptors in immune cells. Although the authors excluded the influence of FAO on ACSL5-mediated MHC-I expression in this paper [<span>7</span>], the metabolic perturbation by ACSL5 is still complicated and further exploration of deeper molecular mechanisms underlying EA-ACSL5 axis in tumor immunology is worthwhile.</p><p>Jiaming Wang and Xuetao Cao drafted the manuscript and figures. Xuetao Cao supervised and revised the manuscript.</p><p>The authors declare that they have no competing interests.</p><p>Not applicable.</p>","PeriodicalId":9495,"journal":{"name":"Cancer Communications","volume":"44 5","pages":"580-583"},"PeriodicalIF":24.9000,"publicationDate":"2024-04-12","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://onlinelibrary.wiley.com/doi/epdf/10.1002/cac2.12543","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Cancer Communications","FirstCategoryId":"3","ListUrlMain":"https://onlinelibrary.wiley.com/doi/10.1002/cac2.12543","RegionNum":1,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"ONCOLOGY","Score":null,"Total":0}
引用次数: 0
Abstract
Overcoming resistance to immune checkpoint blockade (ICB) therapy will pave the way for effective ICB cancer immunotherapy since a large proportion of cancer patients are not responsive to ICB immunotherapy [1]. The molecular mechanisms of ICB resistance are diverse, including tumor-intrinsic resistant factors (such as genetic and epigenetic disorders), immunosuppressive/disabled factors (such as T cell exhaustion), and environmental restrictive factors (such as neuroendocrine stress and metabolic reprogramming). Deciphering the mechanisms for ICB resistance will provide immense potential for designing new immunotherapeutic strategies in refractory cancers. The tumor microenvironment (TME) includes diverse types of cells, such as immune cells, cancer-associated fibroblasts, and endothelial cells, as well as intercellular cytokines/chemokines, growth factors, and metabolites, which have been recognized as crucial determinants in ICB responsiveness. The recent advent of high-throughput metabolomics and lipidomics analysis reveals that metabolic reprogramming in TME is closely associated with cancer cell invasion, regulated cell death, immune escape, and chemoresistance [2]. On the other hand, immunometabolism also modulates tumor-associated immune cell function and immunotherapy efficacy. For instance, higher expression of major facilitator superfamily domain containing 2A (MFSD2A) in gastric cancer cells inhibits transforming growth factor beta 1 (TGF-β1) production by suppressing cyclooxygenase 2 (COX2)-prostaglandin synthesis, thus promoting antitumor immunity via reprogramming TME [3].
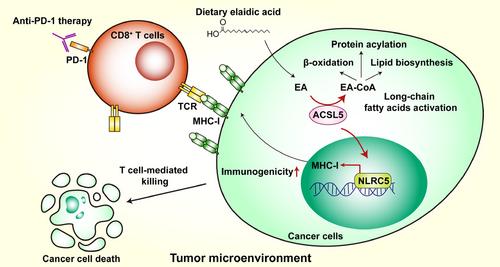
As a major component of lipids in TME, long-chain fatty acids (LCFAs) are important energy supply and cellular membrane components for cancer cells. Indeed, various types of LCFAs have been found in TME, showing different and sometimes opposite influences on tumor progression and antitumor immunity [4]. For example, palmitic acid promotes metastasis in oral carcinomas and melanoma mouse models through stimulating intratumoral Schwann cells and innervation [5], whereas linoleic acid potentiates CD8+ T cell antitumor functions via enhancing endoplasmic reticulum-mitochondria contact formation and energetics fitness [6]. Therefore, better understanding of the molecular mechanism for each individual LCFA in tumor immunity is meaningful since it may improve cancer immunotherapy through targeting metabolic reprogramming of TME. In a recent study published in Cell Metabolism, Lai et al. [7] found dietary elaidic acid (EA) supplementation elevates tumoral major histocompatibility complex-1 (MHC-I) expression via acyl-coenzyme A synthetase long chain family member 5 (ACSL5), thus suppressing tumor growth and enhancing anti-programmed cell death protein 1 (anti-PD-1) efficacy (Figure 1).
Acyl-coenzyme A synthetase long-chain family members (ACSLs) catalyze the conversion of LCFAs into LCFA-coenzyme A (LCFA-CoA) for further lipid catabolism or anabolism. It is reported that interferon gamma (IFNγ) released by cytotoxic T lymphocytes (CTLs) and arachidonic acid in TME coordinately induce tumor cell ferroptosis via acyl-coenzyme A synthetase long chain family member 4 (ACSL4) [8]. Therefore, manipulating ACSLs activity might be an ideal target for metabolic reprogramming in cancer therapy. In this study, the authors found ectopic expression of acyl-coenzyme A synthetase long chain family member 1 (ACSL1), ACSL5, or acyl-coenzyme A synthetase long chain family member (ACSL6) in B16-F10 melanoma cells reduced tumor growth in a subcutaneous mouse model [7]. Given that higher levels of ACSL5 and ACSL6 mRNA were associated with improved overall survival in melanoma patients according to The Cancer Genome Atlas (TCGA) datasets and broad expression of ACSL5 in multiple tissues, the authors focused on ACSL5 for further study. ACSL5 expression also suppressed subcutaneous growth of Lewis lung carcinoma (LLC), Hepa1-6 hepatic tumor, and tumor-model-derived cancer cell line (TDCL) in mouse models. Interestingly, ACSL5-mediated antitumor effect was dependent on acquired immunity since ACSL5 failed to regulate tumor growth in immunodeficient non-obese diabetic-PrkdcscidIL2rgem1/Smoc (M-NSG) and Rag2−/− mice or upon anti-CD8 antibody treatment in C57BL/6 mice. Furthermore, ACSL5-deficient TDCL tumors were not responsive to anti-PD-1 therapy in contrast to wide-type tumors, indicating that tumor cell ACSL5 determines the sensitivity to ICB immunotherapy.
To further delineate ACSL5 antitumor function, the authors examined the number and function of tumor-infiltrating T cells. The number of CD8+ T cells or CD4 + T cells and their cytokines (tumor necrosis factor α [TNF-α] and IFNγ) production were not changed in ACSL5-deficient TDCL tumors. Besides, ACSL5 ectopic B16-F10 tumors had no effects on the number of T cells but slightly increased the TNF-α production by CD8+ T cells. Although CD8+ T cell number was increased in ACSL5-expressing LLC tumors, the number of CD4+ T cells and cytokines production by T cells were not influenced. Therefore, the authors speculated that ACSL5 might regulate tumor sensitivity to T cell-mediated killing. Indeed, they found that ACSL5 deficiency rendered tumor cells more resistant to CD8+ T cell-mediated cytotoxicity in the co-culture system in vitro. Insufficient tumor immunogenicity is a pivotal cause of ICB resistance since CTLs mediate tumor cell killing through T cell receptor (TCR) recognition of tumor antigen presentation by MHC-I molecules [9]. Accordingly, the authors found downregulation of MHC-I expression in ACSL5-deficient tumor cells. Moreover, knockout of β-2 microglobulin (β2m), a component of MHC-I, promoted tumor growth in control tumors, but not in ACSL5-deficient tumors in vivo, indicating ACSL5 mediates antitumor effect through MHC-I upregulation.
By transcriptomic analysis of wild-type and ACSL5-deficient cells, the authors noticed that NLR family CARD domain containing 5 (NLRC5), one of the key transcription activators of MHC-I, was reduced upon ACSL5 knockout. Silencing NLRC5 completely abolished the MHC-I upregulation by ACSL5 ectopic expression, suggesting the ACSL5-NLRC5-MHC-I axis in determining tumor cell immunogenicity for induction of antitumor immunity.
Given that ACSLs take part in LCFAs activation, whether any LCFAs supplementation phenocopies ACSL5's antitumor function? According to various LCFAs screening, the authors found EA, an oleic acid trans isomer and a common component of animal fats and vegetable oils, potently enhanced basal and IFNγ-stimulated MHC-I expression. ACSL5 or NLRC5 deficiency impaired the induction of MHC-I by EA, further validating the EA-ACSL5-NLRC5-MHC-I axis in metabolic regulation of antitumor immunity. Besides, intraperitoneal and intragastrical EA administration both suppressed B16-F10 tumor growth in vivo in an immune-dependent manner. Consistently, EA treatment in combination with anti-PD-1 therapy significantly reduced LLC tumor burden, which was resistant to ICB originally, underscoring the promising therapeutic potential of dietary EA supplementation in promoting responsiveness to ICB. Finally, the authors found that higher plasma EA level was correlated with patients with better response to ICB and improved progression-free survival, implying that plasma EA might be an ideal predictor for ICB effectiveness.
Although numerous studies suggest the harmful effect of trans fatty acids (TFAs) especially in cardiovascular diseases, this study unveiled an unexpected role of TFAs in boosting tumor immunogenicity and enhancing antitumor immune response. However, there are still many questions that need to be addressed. How the metabolic enzyme ACSL5 regulates nuclear transcription factor NLRC5 expression, and how NLRC5 acts as a fatty acid metabolism alteration sensor are unclear. In contrast to this finding, EA has been reported to promote the growth and metastasis of mouse colorectal cancer cells by activating Wnt and extracellular signal-regulated kinase (ERK) signaling [10]. Whether the opposite functions of EA in different tumor models are due to TME heterogeneity needs further investigation. Although the authors focused on the antitumor effect of EA in tumor cells, whether EA also directly affects CD8+ T cells function is unclear. It is also interesting to examine the lipid metabolism rewiring, fatty acid-mediated signaling transduction and antitumor function modulation in immune cells during EA intake. The authors demonstrated that ACSL5 converted EA into EA-CoA using isotype labeling, however, whether EA-CoA stimulated MHC-I expression directly through regulating target protein acylation like palmitoylation remains elusive. After conversion to EA-CoA, the activated fatty acids were essential for the downstream lipid metabolic pathways, such as fatty acid β-oxidation (FAO), lipid biosynthesis, and protein acylation (Figure 1). Our unpublished data suggest that ACSL5 can regulate phospholipid synthesis and cellular membrane properties, which might also impact membrane translocation of MHC-1 in tumor cells and other receptors in immune cells. Although the authors excluded the influence of FAO on ACSL5-mediated MHC-I expression in this paper [7], the metabolic perturbation by ACSL5 is still complicated and further exploration of deeper molecular mechanisms underlying EA-ACSL5 axis in tumor immunology is worthwhile.
Jiaming Wang and Xuetao Cao drafted the manuscript and figures. Xuetao Cao supervised and revised the manuscript.
The authors declare that they have no competing interests.
期刊介绍:
Cancer Communications is an open access, peer-reviewed online journal that encompasses basic, clinical, and translational cancer research. The journal welcomes submissions concerning clinical trials, epidemiology, molecular and cellular biology, and genetics.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
