Razieh Khoshnevisan, Shakiba Hassanzadeh, Christoph Klein, Meino Rohlfs, Bodo Grimbacher, Newsha Molavi, Aryana Zamanifar, Ali Khoshnevisan, Mahbube Jafari, Bahram Bagherpour, Mahdiyeh Behnam, Somayeh Najafi, Roya Sherkat
{"title":"B-cells absence in patients diagnosed as inborn errors of immunity: a registry-based study","authors":"Razieh Khoshnevisan, Shakiba Hassanzadeh, Christoph Klein, Meino Rohlfs, Bodo Grimbacher, Newsha Molavi, Aryana Zamanifar, Ali Khoshnevisan, Mahbube Jafari, Bahram Bagherpour, Mahdiyeh Behnam, Somayeh Najafi, Roya Sherkat","doi":"10.1007/s00251-024-01342-y","DOIUrl":null,"url":null,"abstract":"<p>Hypogammaglobulinemia without B-cells is a subgroup of inborn errors of immunity (IEI) which is characterized by a significant decline in all serum immunoglobulin isotypes, coupled with a pronounced reduction or absence of B-cells. Approximately 80 to 90% of individuals exhibit genetic variations in Bruton’s agammaglobulinemia tyrosine kinase (BTK), whereas a minority of cases, around 5–10%, are autosomal recessive agammaglobulinemia (ARA). Very few cases are grouped into distinct subcategories. We evaluated phenotypically and genetically 27 patients from 13 distinct families with hypogammaglobinemia and no B-cells. Genetic analysis was performed via whole-exome and Sanger sequencing. The most prevalent genetic cause was mutations in <i>BTK</i>. Three novel mutations in the <i>BTK</i> gene include c.115 T > C (p. Tyr39His), c.685-686insTTAC (p.Asn229llefs5), and c.163delT (p.Ser55GlnfsTer2). Our three ARA patients include a novel homozygous stop-gain mutation in the immunoglobulin heavy constant Mu chain (<i>IGHM</i>) gene, a novel frameshift mutation of the B-cell antigen receptor complex-associated protein (<i>CD79A</i>) gene, a novel bi-allelic stop-gain mutation in the transcription factor 3 (<i>TCF3</i>) gene. Three patients with agammaglobulinemia have an autosomal dominant inheritance pattern, which includes a missense variant in <i>PIK3CD</i>, a novel missense variant in <i>PIK3R1</i> and a homozygous silent mutation in the phosphoinositide-3-kinase regulatory subunit (<i>RASGRP1</i>) gene. This study broadens the genetic spectrum of hypogammaglobulinemia without B-cells and presented a few novel variants within the Iranian community, which may also have implications in other Middle Eastern populations. Notably, disease control was better in the second affected family member in families with multiple cases.</p>","PeriodicalId":13446,"journal":{"name":"Immunogenetics","volume":"73 1","pages":""},"PeriodicalIF":2.9000,"publicationDate":"2024-04-29","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Immunogenetics","FirstCategoryId":"3","ListUrlMain":"https://doi.org/10.1007/s00251-024-01342-y","RegionNum":4,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"GENETICS & HEREDITY","Score":null,"Total":0}
引用次数: 0
Abstract
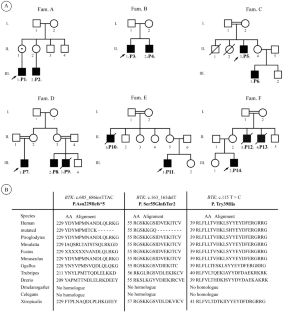
Hypogammaglobulinemia without B-cells is a subgroup of inborn errors of immunity (IEI) which is characterized by a significant decline in all serum immunoglobulin isotypes, coupled with a pronounced reduction or absence of B-cells. Approximately 80 to 90% of individuals exhibit genetic variations in Bruton’s agammaglobulinemia tyrosine kinase (BTK), whereas a minority of cases, around 5–10%, are autosomal recessive agammaglobulinemia (ARA). Very few cases are grouped into distinct subcategories. We evaluated phenotypically and genetically 27 patients from 13 distinct families with hypogammaglobinemia and no B-cells. Genetic analysis was performed via whole-exome and Sanger sequencing. The most prevalent genetic cause was mutations in BTK. Three novel mutations in the BTK gene include c.115 T > C (p. Tyr39His), c.685-686insTTAC (p.Asn229llefs5), and c.163delT (p.Ser55GlnfsTer2). Our three ARA patients include a novel homozygous stop-gain mutation in the immunoglobulin heavy constant Mu chain (IGHM) gene, a novel frameshift mutation of the B-cell antigen receptor complex-associated protein (CD79A) gene, a novel bi-allelic stop-gain mutation in the transcription factor 3 (TCF3) gene. Three patients with agammaglobulinemia have an autosomal dominant inheritance pattern, which includes a missense variant in PIK3CD, a novel missense variant in PIK3R1 and a homozygous silent mutation in the phosphoinositide-3-kinase regulatory subunit (RASGRP1) gene. This study broadens the genetic spectrum of hypogammaglobulinemia without B-cells and presented a few novel variants within the Iranian community, which may also have implications in other Middle Eastern populations. Notably, disease control was better in the second affected family member in families with multiple cases.
期刊介绍:
Immunogenetics publishes original papers, brief communications, and reviews on research in the following areas: genetics and evolution of the immune system; genetic control of immune response and disease susceptibility; bioinformatics of the immune system; structure of immunologically important molecules; and immunogenetics of reproductive biology, tissue differentiation, and development.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
