{"title":"Strategies for diagnosis and management of CMMRD in low-resource countries: report of a Tunisian family","authors":"Rania Abdelmaksoud-Dammak, Nihel Ammous-Boukhris, Dorra BenAyed-Guerfali, Yassine Gdoura, Imen Boujelben, Souhir Guidara, Slim Charfi, Wiem Boudabbous, Saloua Ammar, Wiem Rhaiem, Mohamed Zaher Boudawara, Hassen Kamoun, Tahya Sallemi-Boudawara, Riadh Mhiri, Raja Mokdad-Gargouri","doi":"10.1007/s10689-024-00386-z","DOIUrl":null,"url":null,"abstract":"<p>Constitutional Mismatch Repair Deficiency (CMMRD) is a rare childhood cancer predisposition syndrome, caused by biallelic pathogenic germline variants in the mismatch repair genes. Diagnosis and management of this syndrome is challenging, especially in low-resource settings. This study describes a patient diagnosed with colorectal cancer and grade 3 astrocytoma at the age of 11 and 12 respectively. Immunohistochemistry analysis showed a loss of MSH2 and MSH6 protein expression in CRC tissues of the patient. We identified by Targeted Exome Sequencing a homozygous pathogenic germline variant in exon 9 of the <i>MSH6</i> gene (c.3991 C > T; p.Ala1268Glyfs*6). Genetic investigation of the family showed that the father was heterozygous for the identified pathogenic variant while the brother was wild type for this variant. Our study highlights the importance of a correct and timely diagnosis of CMMRD which can have implications for treatment. It also underlines the imperative need to enhance awareness, diagnostic standards, and surveillance that are crucial for patients and their families.</p>","PeriodicalId":12336,"journal":{"name":"Familial Cancer","volume":"11 1","pages":""},"PeriodicalIF":2.0000,"publicationDate":"2024-04-30","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Familial Cancer","FirstCategoryId":"3","ListUrlMain":"https://doi.org/10.1007/s10689-024-00386-z","RegionNum":4,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q3","JCRName":"GENETICS & HEREDITY","Score":null,"Total":0}
引用次数: 0
Abstract
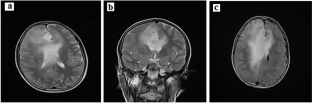
Constitutional Mismatch Repair Deficiency (CMMRD) is a rare childhood cancer predisposition syndrome, caused by biallelic pathogenic germline variants in the mismatch repair genes. Diagnosis and management of this syndrome is challenging, especially in low-resource settings. This study describes a patient diagnosed with colorectal cancer and grade 3 astrocytoma at the age of 11 and 12 respectively. Immunohistochemistry analysis showed a loss of MSH2 and MSH6 protein expression in CRC tissues of the patient. We identified by Targeted Exome Sequencing a homozygous pathogenic germline variant in exon 9 of the MSH6 gene (c.3991 C > T; p.Ala1268Glyfs*6). Genetic investigation of the family showed that the father was heterozygous for the identified pathogenic variant while the brother was wild type for this variant. Our study highlights the importance of a correct and timely diagnosis of CMMRD which can have implications for treatment. It also underlines the imperative need to enhance awareness, diagnostic standards, and surveillance that are crucial for patients and their families.
期刊介绍:
In recent years clinical cancer genetics has become increasingly important. Several events, in particular the developments in DNA-based technology, have contributed to this evolution. Clinical cancer genetics has now matured to a medical discipline which is truly multidisciplinary in which clinical and molecular geneticists work together with clinical and medical oncologists as well as with psycho-social workers.
Due to the multidisciplinary nature of clinical cancer genetics most papers are currently being published in a wide variety of journals on epidemiology, oncology and genetics. Familial Cancer provides a forum bringing these topics together focusing on the interests and needs of the clinician.
The journal mainly concentrates on clinical cancer genetics. Most major areas in the field shall be included, such as epidemiology of familial cancer, molecular analysis and diagnosis, clinical expression, treatment and prevention, counselling and the health economics of familial cancer.




 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
