Sarah Lerario, Luigi Monti, Irene Ambrosetti, Agnese Luglio, Andrea Pietra, Valeria Aiello, Francesca Montanari, Antonio Bellasi, Gianluigi Zaza, Antonio Galante, Davide Salera, Irene Capelli, Gaetano La Manna, Michele Provenzano
{"title":"Fabry disease: a rare disorder calling for personalized medicine.","authors":"Sarah Lerario, Luigi Monti, Irene Ambrosetti, Agnese Luglio, Andrea Pietra, Valeria Aiello, Francesca Montanari, Antonio Bellasi, Gianluigi Zaza, Antonio Galante, Davide Salera, Irene Capelli, Gaetano La Manna, Michele Provenzano","doi":"10.1007/s11255-024-04042-4","DOIUrl":null,"url":null,"abstract":"<p><p>Fabry Disease (FD) is a genetic disease caused by a deficiency in the activity of lysosomal galactosidase A (α-GalA), an enzyme responsible for the catabolism of globotriaosylceramide (Gb3). Since lysosomes are present throughout the body and play a crucial role in catabolism and recycling of cytosolic compounds, FD can affect multiple organs and result in various symptoms, including renal, cardiovascular, neurological, cutaneous, and ophthalmic manifestations. Due to the nonspecific symptoms and the rarity of FD, it is often diagnosed late in life. However, introducing targeted therapies such as enzyme replacement therapy (ERT) and chaperone therapy has significantly improved FD's natural history and prognosis by restoring α-GalA enzyme activity. Despite the advancements, there are limitations to the currently available therapies, which has prompted research into new potential treatments for FD, including alternative forms of enzyme replacement therapy, substrate reduction therapy, mRNA therapy, and genetic therapy. In this review, we analyze the epidemiology, pathophysiology, and treatment of FD, with particular emphasis on promising therapeutic opportunities that could shift the treatment of this rare disease from a standardized to a personalized approach soon.</p>","PeriodicalId":14454,"journal":{"name":"International Urology and Nephrology","volume":" ","pages":"3161-3172"},"PeriodicalIF":1.9000,"publicationDate":"2024-10-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11405476/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"International Urology and Nephrology","FirstCategoryId":"3","ListUrlMain":"https://doi.org/10.1007/s11255-024-04042-4","RegionNum":4,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2024/4/13 0:00:00","PubModel":"Epub","JCR":"Q3","JCRName":"UROLOGY & NEPHROLOGY","Score":null,"Total":0}
引用次数: 0
Abstract
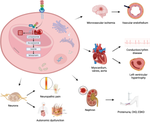
Fabry Disease (FD) is a genetic disease caused by a deficiency in the activity of lysosomal galactosidase A (α-GalA), an enzyme responsible for the catabolism of globotriaosylceramide (Gb3). Since lysosomes are present throughout the body and play a crucial role in catabolism and recycling of cytosolic compounds, FD can affect multiple organs and result in various symptoms, including renal, cardiovascular, neurological, cutaneous, and ophthalmic manifestations. Due to the nonspecific symptoms and the rarity of FD, it is often diagnosed late in life. However, introducing targeted therapies such as enzyme replacement therapy (ERT) and chaperone therapy has significantly improved FD's natural history and prognosis by restoring α-GalA enzyme activity. Despite the advancements, there are limitations to the currently available therapies, which has prompted research into new potential treatments for FD, including alternative forms of enzyme replacement therapy, substrate reduction therapy, mRNA therapy, and genetic therapy. In this review, we analyze the epidemiology, pathophysiology, and treatment of FD, with particular emphasis on promising therapeutic opportunities that could shift the treatment of this rare disease from a standardized to a personalized approach soon.
期刊介绍:
International Urology and Nephrology publishes original papers on a broad range of topics in urology, nephrology and andrology. The journal integrates papers originating from clinical practice.




 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
