Lajoyce Mboning, Liudmilla Rubbi, Michael Thompson, Louis-S Bouchard, Matteo Pellegrini
{"title":"BayesAge: A maximum likelihood algorithm to predict epigenetic age.","authors":"Lajoyce Mboning, Liudmilla Rubbi, Michael Thompson, Louis-S Bouchard, Matteo Pellegrini","doi":"10.3389/fbinf.2024.1329144","DOIUrl":null,"url":null,"abstract":"<p><p><b>Introduction:</b> DNA methylation, specifically the formation of 5-methylcytosine at the C5 position of cytosine, undergoes reproducible changes as organisms age, establishing it as a significant biomarker in aging studies. Epigenetic clocks, which integrate methylation patterns to predict age, often employ linear models based on penalized regression, yet they encounter challenges in handling missing data, count-based bisulfite sequence data, and interpretation. <b>Methods:</b> To address these limitations, we introduce BayesAge, an extension of the scAge methodology originally designed for single-cell DNA methylation analysis. BayesAge employs maximum likelihood estimation (MLE) for age inference, models count data using binomial distributions, and incorporates LOWESS smoothing to capture non-linear methylation-age dynamics. This approach is tailored for bulk bisulfite sequencing datasets. <b>Results:</b> BayesAge demonstrates superior performance compared to scAge. Notably, its age residuals exhibit no age association, offering a less biased representation of epigenetic age variation across populations. Furthermore, BayesAge facilitates the estimation of error bounds on age inference. When applied to down-sampled data, BayesAge achieves a higher coefficient of determination between predicted and actual ages compared to both scAge and penalized regression. <b>Discussion:</b> BayesAge presents a promising advancement in epigenetic age prediction, addressing key challenges encountered by existing models. By integrating robust statistical techniques and tailored methodologies for count-based data, BayesAge offers improved accuracy and interpretability in predicting age from bulk bisulfite sequencing datasets. Its ability to estimate error bounds enhances the reliability of age inference, thereby contributing to a more comprehensive understanding of epigenetic aging processes.</p>","PeriodicalId":73066,"journal":{"name":"Frontiers in bioinformatics","volume":"4 ","pages":"1329144"},"PeriodicalIF":3.6000,"publicationDate":"2024-04-04","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11024280/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Frontiers in bioinformatics","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.3389/fbinf.2024.1329144","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2024/1/1 0:00:00","PubModel":"eCollection","JCR":"Q2","JCRName":"MATHEMATICAL & COMPUTATIONAL BIOLOGY","Score":null,"Total":0}
引用次数: 0
Abstract
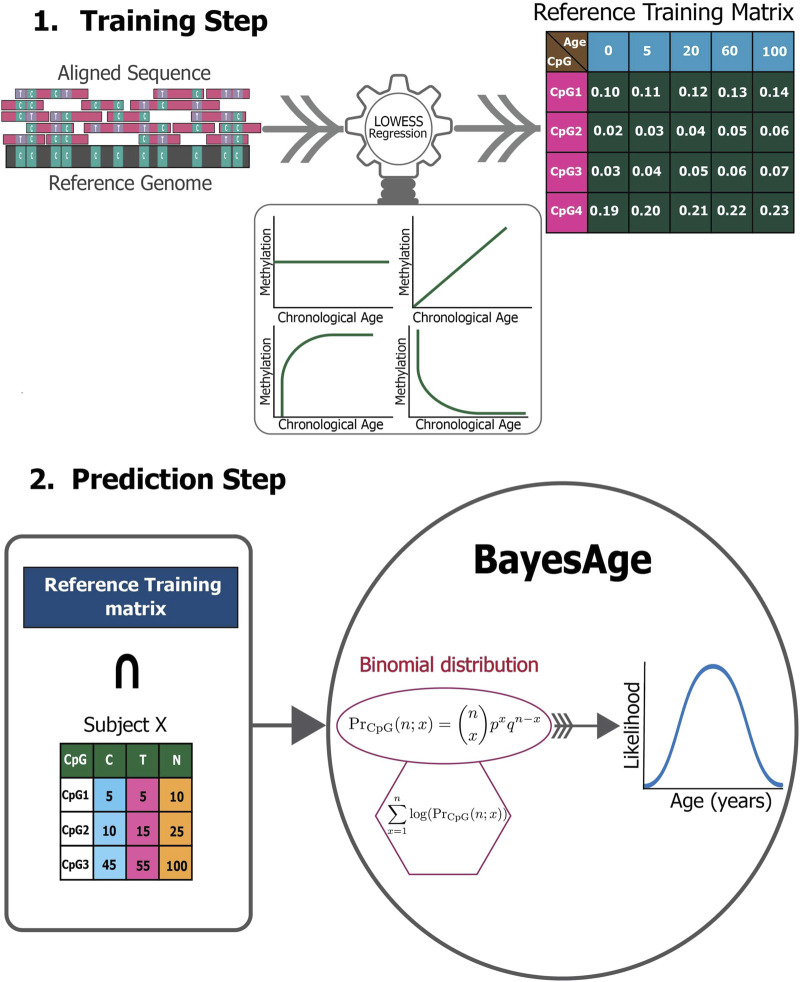
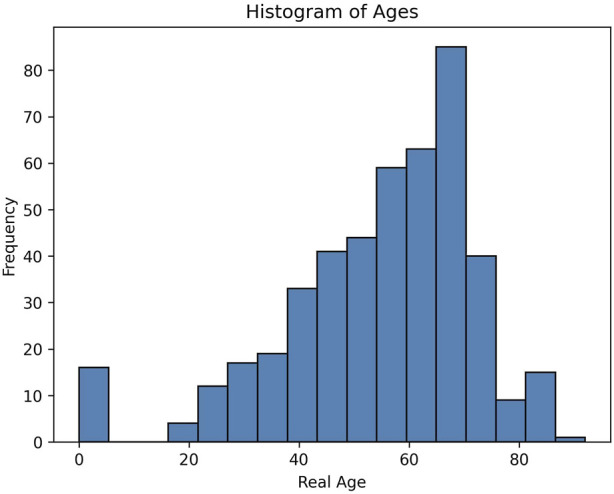
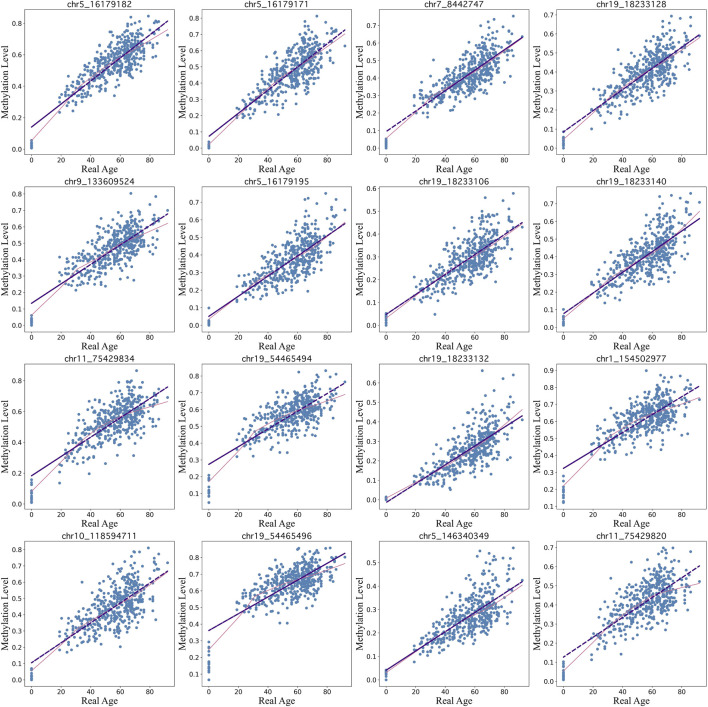
Introduction: DNA methylation, specifically the formation of 5-methylcytosine at the C5 position of cytosine, undergoes reproducible changes as organisms age, establishing it as a significant biomarker in aging studies. Epigenetic clocks, which integrate methylation patterns to predict age, often employ linear models based on penalized regression, yet they encounter challenges in handling missing data, count-based bisulfite sequence data, and interpretation. Methods: To address these limitations, we introduce BayesAge, an extension of the scAge methodology originally designed for single-cell DNA methylation analysis. BayesAge employs maximum likelihood estimation (MLE) for age inference, models count data using binomial distributions, and incorporates LOWESS smoothing to capture non-linear methylation-age dynamics. This approach is tailored for bulk bisulfite sequencing datasets. Results: BayesAge demonstrates superior performance compared to scAge. Notably, its age residuals exhibit no age association, offering a less biased representation of epigenetic age variation across populations. Furthermore, BayesAge facilitates the estimation of error bounds on age inference. When applied to down-sampled data, BayesAge achieves a higher coefficient of determination between predicted and actual ages compared to both scAge and penalized regression. Discussion: BayesAge presents a promising advancement in epigenetic age prediction, addressing key challenges encountered by existing models. By integrating robust statistical techniques and tailored methodologies for count-based data, BayesAge offers improved accuracy and interpretability in predicting age from bulk bisulfite sequencing datasets. Its ability to estimate error bounds enhances the reliability of age inference, thereby contributing to a more comprehensive understanding of epigenetic aging processes.



 分享
分享

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
