{"title":"Review of Phenotypic Heterogeneity of Neuronal Intranuclear Inclusion Disease and <i>NOTCH2NLC</i>-Related GGC Repeat Expansion Disorders.","authors":"Tao Zhang, Lei Bao, Hao Chen","doi":"10.1212/NXG.0000000000200132","DOIUrl":null,"url":null,"abstract":"<p><p>Neuronal intranuclear inclusion disease (NIID) is an underdiagnosed neurodegenerative disorder caused by pathogenic GGC expansions in <i>NOTCH2NLC</i>. However, an increasing number of reports of <i>NOTCH2NLC</i> GGC expansions in patients with Alzheimer disease, essential tremor, Parkinson disease, amyotrophic lateral sclerosis, and oculopharyngodistal myopathy have led to the proposal of a new concept known as <i>NOTCH2NLC</i>-related GGC repeat expansion disorders (NREDs). The majority of studies have mainly focused on screening for <i>NOTCH2NLC</i> GGC repeat variation in populations previously diagnosed with the associated disease, subsequently presenting it as a novel causative gene for the condition. These studies appear to be clinically relevant but do have their limitations because they may incorrectly regard the lack of MRI abnormalities as an exclusion criterion for NIID or overlook concomitant clinical presentations not typically observed in the associated diseases. Besides, in many instances within these reports, patients lack pathologic evidence or undergo long-term follow-up to conclusively rule out NIID. In this review, we will systematically review the research on <i>NOTCH2NLC</i> 5' untranslated region GGC repeat expansions and their association with related neurologic disorders, explaining the limitations of the relevant reports. Furthermore, we will integrate subsequent studies to further demonstrate that these patients actually experienced distinct clinical phenotypes of NIID.</p>","PeriodicalId":48613,"journal":{"name":"Neurology-Genetics","volume":"10 2","pages":"e200132"},"PeriodicalIF":3.7000,"publicationDate":"2024-04-03","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC10997217/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Neurology-Genetics","FirstCategoryId":"3","ListUrlMain":"https://doi.org/10.1212/NXG.0000000000200132","RegionNum":3,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2024/4/1 0:00:00","PubModel":"eCollection","JCR":"Q2","JCRName":"CLINICAL NEUROLOGY","Score":null,"Total":0}
引用次数: 0
Abstract
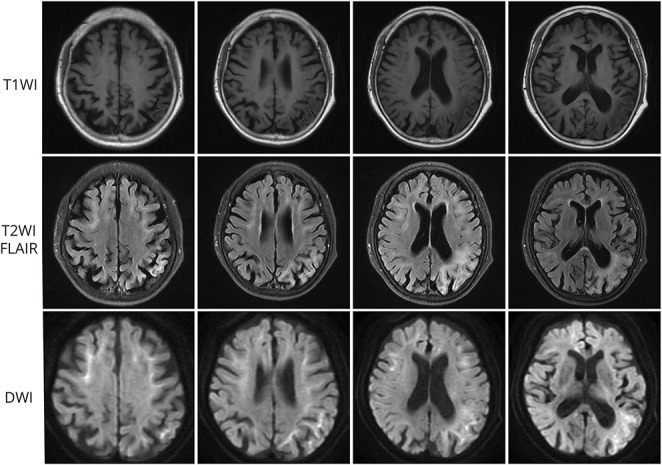
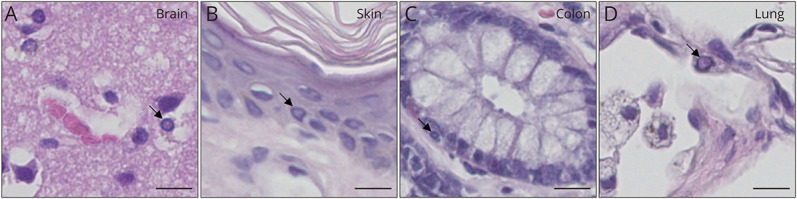
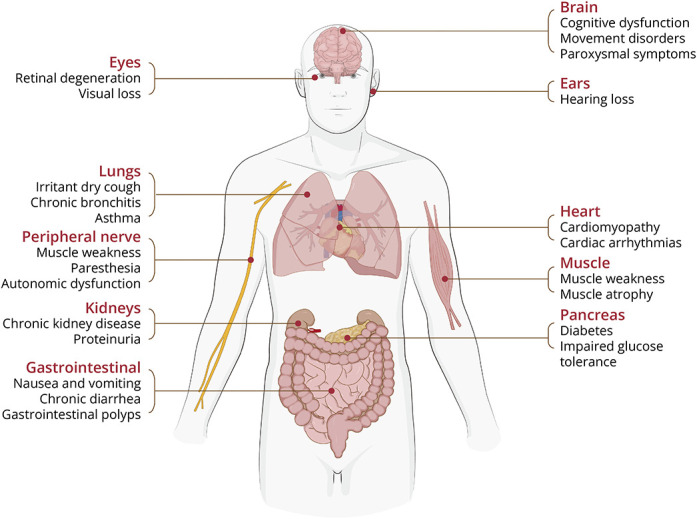
Neuronal intranuclear inclusion disease (NIID) is an underdiagnosed neurodegenerative disorder caused by pathogenic GGC expansions in NOTCH2NLC. However, an increasing number of reports of NOTCH2NLC GGC expansions in patients with Alzheimer disease, essential tremor, Parkinson disease, amyotrophic lateral sclerosis, and oculopharyngodistal myopathy have led to the proposal of a new concept known as NOTCH2NLC-related GGC repeat expansion disorders (NREDs). The majority of studies have mainly focused on screening for NOTCH2NLC GGC repeat variation in populations previously diagnosed with the associated disease, subsequently presenting it as a novel causative gene for the condition. These studies appear to be clinically relevant but do have their limitations because they may incorrectly regard the lack of MRI abnormalities as an exclusion criterion for NIID or overlook concomitant clinical presentations not typically observed in the associated diseases. Besides, in many instances within these reports, patients lack pathologic evidence or undergo long-term follow-up to conclusively rule out NIID. In this review, we will systematically review the research on NOTCH2NLC 5' untranslated region GGC repeat expansions and their association with related neurologic disorders, explaining the limitations of the relevant reports. Furthermore, we will integrate subsequent studies to further demonstrate that these patients actually experienced distinct clinical phenotypes of NIID.
期刊介绍:
Neurology: Genetics is an online open access journal publishing peer-reviewed reports in the field of neurogenetics. Original articles in all areas of neurogenetics will be published including rare and common genetic variation, genotype-phenotype correlations, outlier phenotypes as a result of mutations in known disease-genes, and genetic variations with a putative link to diseases. This will include studies reporting on genetic disease risk and pharmacogenomics. In addition, Neurology: Genetics will publish results of gene-based clinical trials (viral, ASO, etc.). Genetically engineered model systems are not a primary focus of Neurology: Genetics, but studies using model systems for treatment trials are welcome, including well-powered studies reporting negative results.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
