Vidal Yahya, Robertino Dilena, Roberto Del Bo, Manuela Magni, Fabio Biella, Sabrina Salani, Francesco Fortunato, Elisa Scola, Alessio Di Fonzo, Edoardo Monfrini
{"title":"Soft cerebellar signs unveil RARS2-related epilepsy","authors":"Vidal Yahya, Robertino Dilena, Roberto Del Bo, Manuela Magni, Fabio Biella, Sabrina Salani, Francesco Fortunato, Elisa Scola, Alessio Di Fonzo, Edoardo Monfrini","doi":"10.1002/epd2.20237","DOIUrl":null,"url":null,"abstract":"<p>Pathogenic <i>RARS2</i> variants, by critically reducing mitochondrial arginyl-tRNA aminoacylation activity, cause a rare autosomal recessive disease, generally presenting as a severe encephalopathy with onset at birth, premature death, microcephaly, drug-resistant epilepsy, and hypotonia.<span><sup>1-9</sup></span> Epilepsy was reported in ~90% of the cases, often with myoclonic and clonic seizures.<span><sup>2</sup></span> As in other primary mitochondrial diseases, epilepsy may be attributed to ATP depletion, resulting in loss of neuron hyperpolarization (Na<sup>+</sup>/K<sup>+</sup> ATPase activity impairment) and increased excitation (loss of GABA-mediated inhibition).<span><sup>10</sup></span> The most frequent neuroradiological finding is cerebral atrophy, followed by pontocerebellar hypoplasia.<span><sup>1-9</sup></span> We report the clinical and genetic findings of a 16-year-old boy with <i>RARS2</i>-related encephalopathy distinguished for later onset, longer survival, and milder phenotype compared to previously reported cases.</p><p>A 13-year-old Italian boy without family history of neurological diseases was brought to the emergency room for a sleep-related focal-to-bilateral seizure. His mother reported the following ictal semiology: paroxysmal arousal from sleep, sitting up on the bed, head deviation, drooling, grunting, and asymmetric limb tonic posturing with right upper limb extension and left upper limb flexion (figure 4 sign), without awareness nor response to stimuli, with full recovery in ~10 min. EEG showed a 7 Hz background rhythm with brief trains of synchronous and asynchronous frontal 3 Hz sharp waves, prevalent on the right side (Figure 1A). After a similar second seizure on the following day, levetiracetam 500 mg twice a day was started achieving a seizure-free three-year follow-up and a consistent EEG improvement.</p><p>The patient's medical history revealed normal pregnancy, birth, and early motor development with autonomous walking at 15 months. A single episode of febrile seizure at 1 year of age was reported. At 3 years, he was referred to a pediatric neurologist for language delay with increased nonverbal communication; square wave jerks, interrupted pursuit, mild dysmetria, and tandem gait inability were observed at that time. Psychomotor and speech therapy alongside with professional school support were provided with benefit. A brain MRI at the age of 13 years revealed isolated cerebellar vermis atrophy (Figure 1B). At the last neurological examination, at 16 years, the patient displayed mild intellectual disability and cerebellar features including dysarthria, gaze difficulties, postural and kinetic tremor of upper limbs, limbs dysmetria, dysdiadochokinesia, and gait ataxia.</p><p>The combination of focal epilepsy with additional clues including cerebellar ataxia and language delay induced us to perform genetic tests: array-comparative genomic hybridization (array-CGH) resulted negative, whole-exome sequencing revealed two compound heterozygous variants in <i>RARS2</i> (NM_020320.5): c.685C>T (p.Arg229*) and c.972C>T (p.Thr324=) (Figure S1A). The p.Arg229* is a known pathogenic variant.<span><sup>11</sup></span> The c.972C>T, affecting the third to last nucleotide of exon 11, is extremely rare (gnomAD AF = .000004) and predicted in silico to likely impact splicing (SpliceAI = Donor Loss .49/1, dbscSNV ADA score = .919/1, VarSeak = Exon Skipping, class 4/5). Transcript analyses on patient's cDNA demonstrated that the c.972C>T disrupts splicing generating a defective transcript missing exon 11, although allowing the synthesis of some wild-type transcript (Figure 1C), likely able to ensure a significant residual Rars2 enzymatic activity. The presence of this rare splice-disrupting variant in combination with a known <i>RARS2</i> pathogenic variant in a patient with epilepsy and ataxia prompted us to classify it as likely pathogenic (ACMG criteria: PM2, PM3, PS3). Functional analyses on patient's cultured lymphocytes demonstrated a moderate reduction (~39%) of normal-length Rars2 protein compared to two healthy controls (Figure 1D) and a normal mitochondrial respiratory chain activity, consistently with his relatively mild phenotype.</p><p>In conclusion, <i>RARS2</i>-related disease, commonly known as a severe syndromic encephalopathy, may present with more shaded phenotypes representing a diagnostic challenge.<span><sup>3, 5, 6, 9</sup></span> Our patient, distinguished for a mild phenotype, characterized by longer survival, drug-responsive epilepsy, cerebellar ataxia, and mild intellectual disability, highlights that <i>RARS2</i>-encephalopathy should be considered also in older patients evaluated for epilepsy, when subtle additional features like cerebellar ataxia and mild intellectual disability are recorded in medical history. His milder phenotype is likely attributable to a relevant residual wild-type <i>RARS2</i> transcript, supporting the hypothesis that the phenotypic severity is limited by the presence of significant residual functional enzyme. Further research is necessary to delineate a reliable genotype–phenotype correlation for the <i>RARS2</i> gene, to provide patients and their families with improved genetic counseling and clinical assistance.</p><p>None of the authors has any conflict of interest to disclose.</p>","PeriodicalId":50508,"journal":{"name":"Epileptic Disorders","volume":"26 4","pages":"540-543"},"PeriodicalIF":2.7000,"publicationDate":"2024-05-11","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://onlinelibrary.wiley.com/doi/epdf/10.1002/epd2.20237","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Epileptic Disorders","FirstCategoryId":"3","ListUrlMain":"https://onlinelibrary.wiley.com/doi/10.1002/epd2.20237","RegionNum":4,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q3","JCRName":"CLINICAL NEUROLOGY","Score":null,"Total":0}
引用次数: 0
Abstract
Pathogenic RARS2 variants, by critically reducing mitochondrial arginyl-tRNA aminoacylation activity, cause a rare autosomal recessive disease, generally presenting as a severe encephalopathy with onset at birth, premature death, microcephaly, drug-resistant epilepsy, and hypotonia.1-9 Epilepsy was reported in ~90% of the cases, often with myoclonic and clonic seizures.2 As in other primary mitochondrial diseases, epilepsy may be attributed to ATP depletion, resulting in loss of neuron hyperpolarization (Na+/K+ ATPase activity impairment) and increased excitation (loss of GABA-mediated inhibition).10 The most frequent neuroradiological finding is cerebral atrophy, followed by pontocerebellar hypoplasia.1-9 We report the clinical and genetic findings of a 16-year-old boy with RARS2-related encephalopathy distinguished for later onset, longer survival, and milder phenotype compared to previously reported cases.
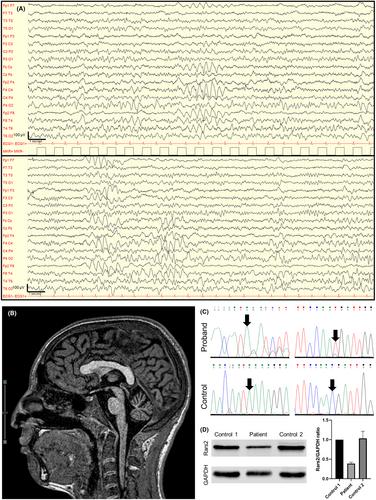
A 13-year-old Italian boy without family history of neurological diseases was brought to the emergency room for a sleep-related focal-to-bilateral seizure. His mother reported the following ictal semiology: paroxysmal arousal from sleep, sitting up on the bed, head deviation, drooling, grunting, and asymmetric limb tonic posturing with right upper limb extension and left upper limb flexion (figure 4 sign), without awareness nor response to stimuli, with full recovery in ~10 min. EEG showed a 7 Hz background rhythm with brief trains of synchronous and asynchronous frontal 3 Hz sharp waves, prevalent on the right side (Figure 1A). After a similar second seizure on the following day, levetiracetam 500 mg twice a day was started achieving a seizure-free three-year follow-up and a consistent EEG improvement.
The patient's medical history revealed normal pregnancy, birth, and early motor development with autonomous walking at 15 months. A single episode of febrile seizure at 1 year of age was reported. At 3 years, he was referred to a pediatric neurologist for language delay with increased nonverbal communication; square wave jerks, interrupted pursuit, mild dysmetria, and tandem gait inability were observed at that time. Psychomotor and speech therapy alongside with professional school support were provided with benefit. A brain MRI at the age of 13 years revealed isolated cerebellar vermis atrophy (Figure 1B). At the last neurological examination, at 16 years, the patient displayed mild intellectual disability and cerebellar features including dysarthria, gaze difficulties, postural and kinetic tremor of upper limbs, limbs dysmetria, dysdiadochokinesia, and gait ataxia.
The combination of focal epilepsy with additional clues including cerebellar ataxia and language delay induced us to perform genetic tests: array-comparative genomic hybridization (array-CGH) resulted negative, whole-exome sequencing revealed two compound heterozygous variants in RARS2 (NM_020320.5): c.685C>T (p.Arg229*) and c.972C>T (p.Thr324=) (Figure S1A). The p.Arg229* is a known pathogenic variant.11 The c.972C>T, affecting the third to last nucleotide of exon 11, is extremely rare (gnomAD AF = .000004) and predicted in silico to likely impact splicing (SpliceAI = Donor Loss .49/1, dbscSNV ADA score = .919/1, VarSeak = Exon Skipping, class 4/5). Transcript analyses on patient's cDNA demonstrated that the c.972C>T disrupts splicing generating a defective transcript missing exon 11, although allowing the synthesis of some wild-type transcript (Figure 1C), likely able to ensure a significant residual Rars2 enzymatic activity. The presence of this rare splice-disrupting variant in combination with a known RARS2 pathogenic variant in a patient with epilepsy and ataxia prompted us to classify it as likely pathogenic (ACMG criteria: PM2, PM3, PS3). Functional analyses on patient's cultured lymphocytes demonstrated a moderate reduction (~39%) of normal-length Rars2 protein compared to two healthy controls (Figure 1D) and a normal mitochondrial respiratory chain activity, consistently with his relatively mild phenotype.
In conclusion, RARS2-related disease, commonly known as a severe syndromic encephalopathy, may present with more shaded phenotypes representing a diagnostic challenge.3, 5, 6, 9 Our patient, distinguished for a mild phenotype, characterized by longer survival, drug-responsive epilepsy, cerebellar ataxia, and mild intellectual disability, highlights that RARS2-encephalopathy should be considered also in older patients evaluated for epilepsy, when subtle additional features like cerebellar ataxia and mild intellectual disability are recorded in medical history. His milder phenotype is likely attributable to a relevant residual wild-type RARS2 transcript, supporting the hypothesis that the phenotypic severity is limited by the presence of significant residual functional enzyme. Further research is necessary to delineate a reliable genotype–phenotype correlation for the RARS2 gene, to provide patients and their families with improved genetic counseling and clinical assistance.
None of the authors has any conflict of interest to disclose.
期刊介绍:
Epileptic Disorders is the leading forum where all experts and medical studentswho wish to improve their understanding of epilepsy and related disorders can share practical experiences surrounding diagnosis and care, natural history, and management of seizures.
Epileptic Disorders is the official E-journal of the International League Against Epilepsy for educational communication. As the journal celebrates its 20th anniversary, it will now be available only as an online version. Its mission is to create educational links between epileptologists and other health professionals in clinical practice and scientists or physicians in research-based institutions. This change is accompanied by an increase in the number of issues per year, from 4 to 6, to ensure regular diffusion of recently published material (high quality Review and Seminar in Epileptology papers; Original Research articles or Case reports of educational value; MultiMedia Teaching Material), to serve the global medical community that cares for those affected by epilepsy.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
