Syed Shah Muhammad, Muhammad Shoaib, Muhammad Tariq Pervez
{"title":"An Integrated Framework for Analysis and Prediction of Impact of Single Nucleotide Polymorphism Associated with Human Diseases.","authors":"Syed Shah Muhammad, Muhammad Shoaib, Muhammad Tariq Pervez","doi":"10.1177/11769343241249916","DOIUrl":null,"url":null,"abstract":"<p><p>Single nucleotide polymorphisms are most common type of genetic variation in human genome. Analyzing genetic variants can help us better understand the genetic basis of diseases and develop predictive models which are useful to identify individuals who are at increased risk for certain diseases. Several SNP analysis tools have already been developed. For running these tools, the user needs to collect data from various databases. Secondly, often researchers have to use multiple variant analysis tools for cross validating their results and increase confidence in their findings. Extracting data from multiple databases and running multiple tools at a time, increases complexity and time required for analysis. There are some web-based tools that integrate multiple genetic variant databases and provide variant annotations for a few tools. These approaches have some limitations such as retrieving annotation information, filtering common pathogenic variants. The proposed web-based tool, namely IPSNP: An Integrated Platform for Predicting Impact of SNPs is written in Django which is a python-based framework. It uses RESTful API of MyVariant.info to extract annotation information of variants associated with a given gene, rsID, HGVS format variants specified in a VCF file for 29 tools. The results are in the form of a CSV file of predictions (1) derived from the consensus decision, (2) a file having annotations for the variants associated with the given gene, (3) a file showing variants declared as pathogenic commonly by the selected tools, and (4) a CSV file containing chromosome coordinates based on GRCh37 and GRCh38 genome assemblies, rsIDs and proteomic data, so that users may use tools of their choice and avoiding manual parameter collection for each tool. IPSNP is a valuable resource for researchers and clinicians and it can help to save time and effort in discovering the novel disease-associated variants and the development of personalized treatments.</p>","PeriodicalId":50472,"journal":{"name":"Evolutionary Bioinformatics","volume":"20 ","pages":"11769343241249916"},"PeriodicalIF":1.5000,"publicationDate":"2024-05-10","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11088291/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Evolutionary Bioinformatics","FirstCategoryId":"99","ListUrlMain":"https://doi.org/10.1177/11769343241249916","RegionNum":4,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2024/1/1 0:00:00","PubModel":"eCollection","JCR":"Q4","JCRName":"EVOLUTIONARY BIOLOGY","Score":null,"Total":0}
引用次数: 0
Abstract
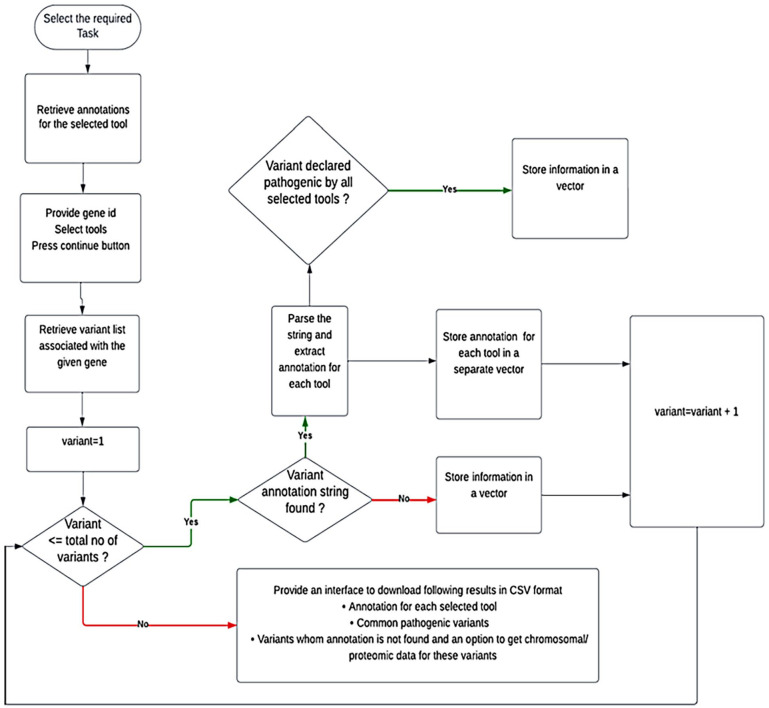
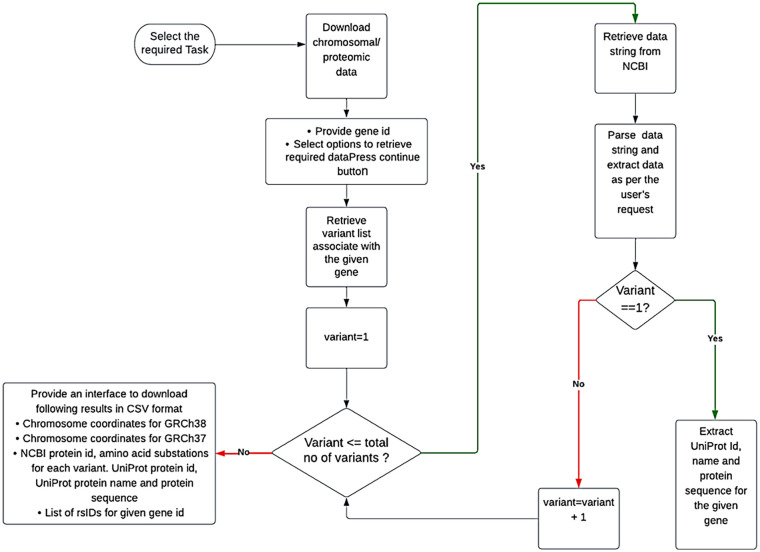
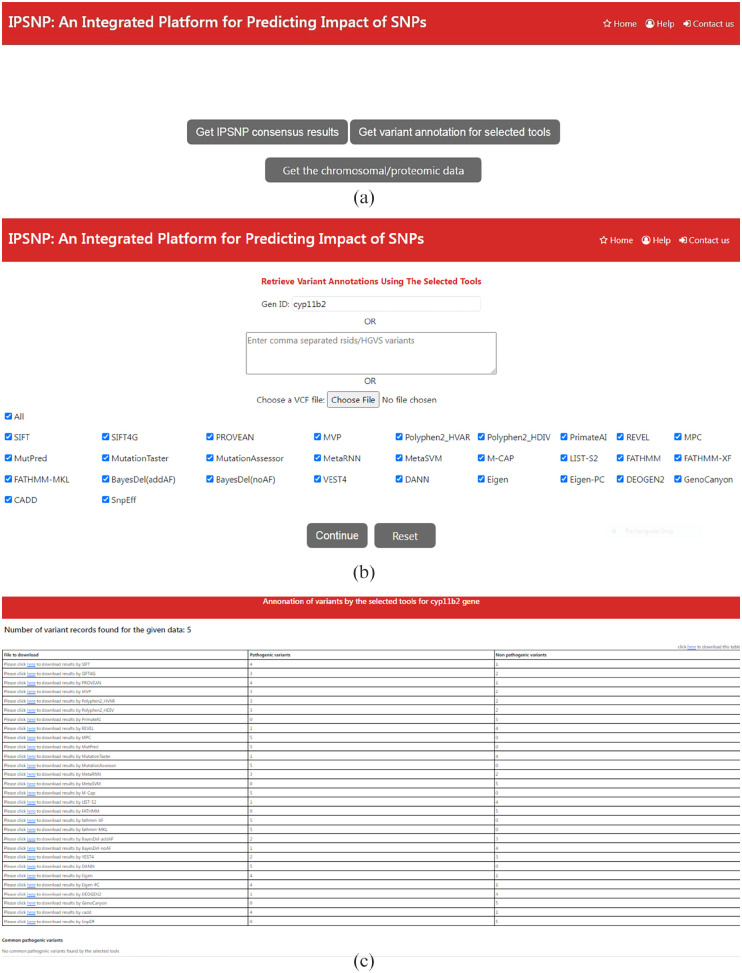
Single nucleotide polymorphisms are most common type of genetic variation in human genome. Analyzing genetic variants can help us better understand the genetic basis of diseases and develop predictive models which are useful to identify individuals who are at increased risk for certain diseases. Several SNP analysis tools have already been developed. For running these tools, the user needs to collect data from various databases. Secondly, often researchers have to use multiple variant analysis tools for cross validating their results and increase confidence in their findings. Extracting data from multiple databases and running multiple tools at a time, increases complexity and time required for analysis. There are some web-based tools that integrate multiple genetic variant databases and provide variant annotations for a few tools. These approaches have some limitations such as retrieving annotation information, filtering common pathogenic variants. The proposed web-based tool, namely IPSNP: An Integrated Platform for Predicting Impact of SNPs is written in Django which is a python-based framework. It uses RESTful API of MyVariant.info to extract annotation information of variants associated with a given gene, rsID, HGVS format variants specified in a VCF file for 29 tools. The results are in the form of a CSV file of predictions (1) derived from the consensus decision, (2) a file having annotations for the variants associated with the given gene, (3) a file showing variants declared as pathogenic commonly by the selected tools, and (4) a CSV file containing chromosome coordinates based on GRCh37 and GRCh38 genome assemblies, rsIDs and proteomic data, so that users may use tools of their choice and avoiding manual parameter collection for each tool. IPSNP is a valuable resource for researchers and clinicians and it can help to save time and effort in discovering the novel disease-associated variants and the development of personalized treatments.
期刊介绍:
Evolutionary Bioinformatics is an open access, peer reviewed international journal focusing on evolutionary bioinformatics. The journal aims to support understanding of organismal form and function through use of molecular, genetic, genomic and proteomic data by giving due consideration to its evolutionary context.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
